Typical application scenarios and breakthrough discoveries of DRS technology
Viral and Pathogen Transcriptomics
DRS has emerged as a useful technology for studying viral and pathogen transcriptomes [277], [278], [279]. Unlike conventional cDNA-based sequencing approaches, DRS enables the sequencing of full-length native RNA molecules without RT or amplification, thereby preserving strand information, transcript structure, and RNA modification signatures [189]. These features have facilitated new analyses for understanding the transcriptional complexity, epitranscriptomic regulation, and rapid detection of infectious agents.
Central to these processes is the recognition that RNA modifications constitute an important regulatory layer in both host and viral gene expression (Figure 4) [280]. Nanopore DRS enables native, full-length RNA reads while capturing modification-associated perturbations in ionic current, providing a complementary alternative to short-read, antibody- or chemistry-based mapping that typically fragments molecules and loses isoform linkage [189], [281]. Using DRS, RNA modifications have been extensively characterized across transcripts from diverse viral genomes, encompassing both DNA viruses (e.g., adenovirus) and RNA viruses such as SARS-CoV-2, HIV, hepatitis B virus (HBV), bamboo mosaic virus (BaMV), and cucumber mosaic virus (CMV) [282], [283]. In 2020, Price et al. utilized nanopore DRS to demonstrate that m6A modifications on adenovirus RNAs are essential for regulating the splicing efficiency of viral late transcripts, highlighting a critical role for epitranscriptomic regulation in viral gene expression [184]. Studies on SARS-CoV-2 virus uncovered that m6A modifications are enriched in the 3′ end of the viral genome [284], [285]. Two studies on HIV have revealed isoform-dependent methylation patterns and unique 2-LTR transcript modifications, and identified several critical m6A sites near the 3’ end of the viral RNA, which are essential regulators of normal HIV-1 RNA splicing and protein translation [286], [287]. Applying nanopore DRS to HBV transcripts, researchers identified four high-confidence m5C sites, demonstrating that HBV mRNAs are extensively m5C-modified and underscoring the power of DRS for precise, transcript-resolved viral epitranscriptome mapping [288]. DRS has also been applied to detect other modifications such as Ψ [289], [290] and Nm [291] on viral RNAs. However, integrative studies combining DRS with complementary approaches have revealed the absence of significant m6A modification sites in certain RNA viruses. This includes mosquito-borne viruses such as chikungunya (CHIKV) and dengue (DENV), as well as plant-infecting viruses like zucchini yellow mosaic virus (ZYMV) and trichosanthes mottle mosaic virus (TrMMV) [292], [293]. Overall, the sites and frequency of RNA modifications in viruses is relatively low, and future studies will require more experiments to further confirm the functions of these potential modifications in virus infection. Besides virus, DRS have also been used to detect RNA modifications in other pathogens. Using modification‑free in vitro transcription (IVT) sample as a negative control, Tan et al. applied nanopore DRS to enable transcriptome‑wide RNA modification detection in Escherichia coli and S. aureus [209].
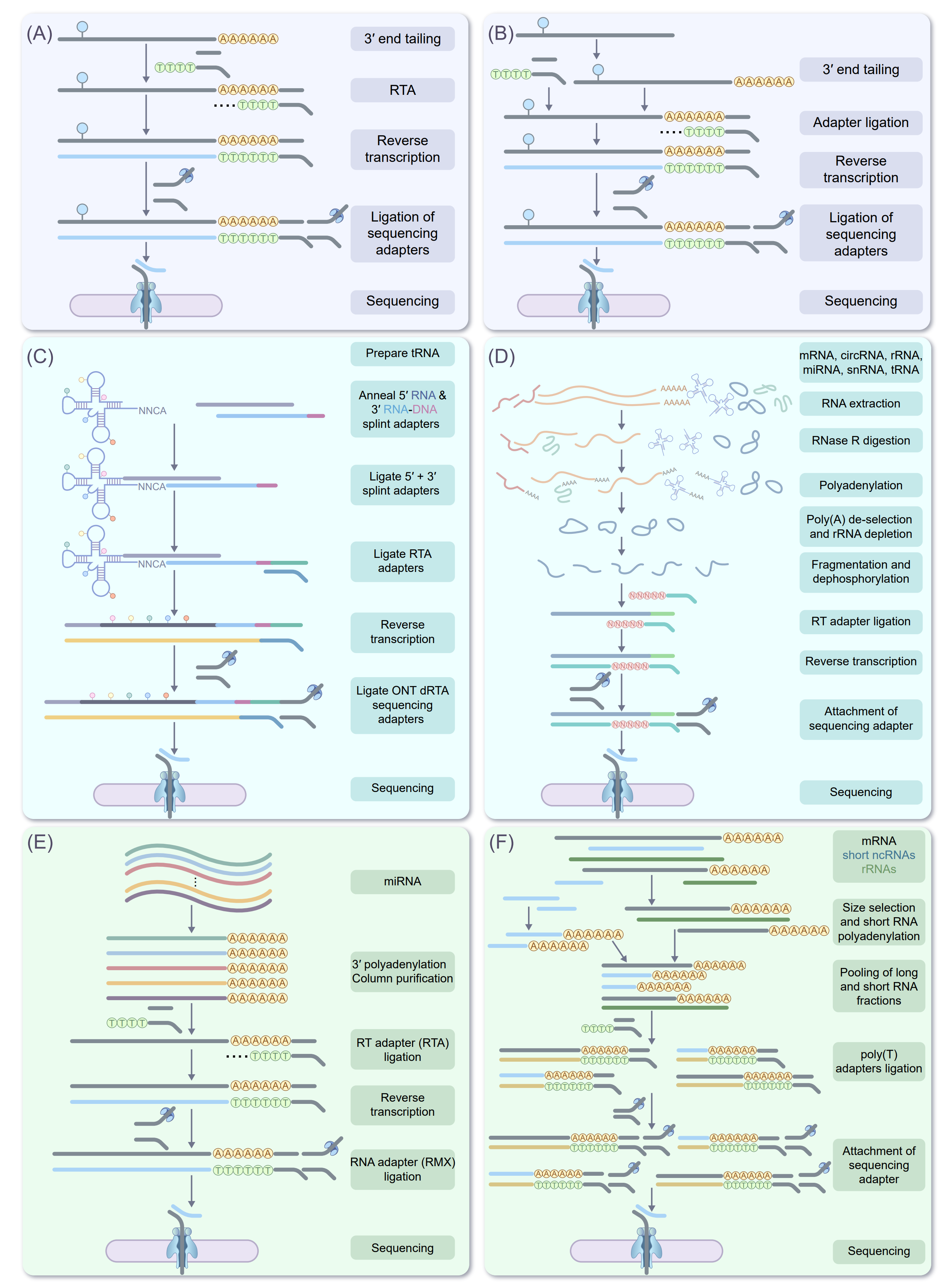
Figure 4. Schematic workflows for DRS of diverse RNA classes. Overview of representative experimental strategies for nanopore‑based direct RNA sequencing of distinct RNA types. (A) mRNA DRS workflow, illustrating 3′‑end tailing (if required), adapter ligation, reverse transcription primed at the 3′ terminus, ligation of sequencing adapters and nanopore sequencing. (B) rRNA workflow, including processing of primary rRNA transcripts, rRNA maturation, and direct RNA approaches, adapter ligation and sequencing. (C) tRNA workflow, showing annealing of 5′ and 3′ splint adapters to mature tRNAs, ligation of reverse‑transcription adapters, cDNA synthesis, ligation of ONT sequencing adapters and subsequent nanopore sequencing. (D) circRNA workflow, involving total RNA extraction, RNase R digestion to enrich circular molecules, polyadenylation and removal of residual linear RNAs, adapter ligation, reverse transcription and loading of circRNA-cDNA hybrids for sequencing. (E) miRNA workflow, in which small RNAs are enriched, subjected to 3′ polyadenylation, ligated to reverse‑transcription and RNA adapters, converted to RNA-cDNA hybrids and sequenced. (F) Non‑poly(A) RNA workflow, depicting parallel processing of long and short non‑polyadenylated transcripts, size selection, pooling, ligation of poly(T) adapters and construction of libraries compatible with DRS.
In addition to epitranscriptomic profiling, the long-read nature of nanopore sequencing enables modification detection in the context of full-length transcripts, allowing researchers to link epitranscriptomic marks with specific isoforms, subgenomic RNAs, or transcriptional variants [287], [294]. This integrative view is particularly valuable for RNA viruses with compact and multifunctional genomes.
Many viruses exhibit highly complex transcriptional strategies, including nested transcription, overlapping open reading frames, and extensive use of subgenomic RNAs [295]. Nanopore DRS with its ability to produce full-length native RNA reads, has substantially advanced the discovery of viral RNA structural variants. For example, nanopore DRS of SARS-CoV-2 revealed a highly complex transcriptome containing numerous previously unannotated subgenomic RNAs, alternative transcription regulatory junctions, and noncanonical fusion transcripts [278], [279]. Similar approaches applied to herpes simplex virus 1 uncovered extensive alternative transcription start and PAS, as well as read-through and antisense transcription, highlighting unexpected transcript isoform diversity during infection [296]. Other researchers have also uncovered previously unannotated transcript isoforms, alternative transcription start and termination sites, and read-through transcription events in diverse RNA viruses [51], [278], [297].
Beyond isoform discovery, DRS enables strand-specific quantification of genomic and antigenomic RNAs, providing insights into replication and transcription dynamics in positive- and negative-sense RNA viruses [278], [296]. In segmented or recombination-prone viruses, long reads help resolve chimeric RNAs and fusion transcripts that may play roles in viral evolution or pathogenicity [296], [298]. Together, these studies demonstrate that nanopore DRS is particularly suited to resolving the structural complexity of viral transcriptomes, providing an integrated view of RNA architecture that is often inaccessible to short-read sequencing technologies. In many clinical or environmental samples, pathogen-derived RNA represents only a small fraction of total RNA, posing challenges for direct sequencing [299], [300]. To address this, targeted and enrichment strategies have been developed to enhance the representation of pathogen transcripts in DRS libraries.
Host RNA depletion strategies, such as ribosomal RNA removal, poly(A) selection, and size selection, can substantially reduce the overwhelming background of host-derived transcripts, thereby increasing the relative abundance of pathogen RNAs in sequencing libraries [209], [301]. Adaptive sampling provides an additional, software-controlled enrichment approach unique to nanopore sequencing, in which short stretches of signal from a molecule are analyzed in real time and compared to reference sequences; reads matching target pathogen genomes are accepted and allowed to continue sequencing, whereas non-target (e.g., host) molecules are rejected by reversing the voltage and ejecting them from the pore, thereby enriching pathogen-derived transcripts during the run [302]. Experimental evaluations using synthetic mock communities demonstrated enrichment of rare species by up to ~14-fold under optimal long-read conditions, with an effective enrichment of ~5-fold after accounting for yield losses due to read rejection [303]. This allows selective enrichment of pathogen-derived molecules during the sequencing run itself, improving efficiency and reducing sequencing costs. Except for ONT’s built-in adaptive sampling strategy, several computational tools have been developed to implement adaptive sampling in real time [304], [305]. Signal-based methods such as UNCALLED [306] and Sigmap [307] directly compare raw current traces with reference-derived signal models to rapidly classify molecules before full sequencing. Sequence-based approaches including Readfish [308], ReadBouncer [309], and RUBRIC [310] first perform ultra-fast basecalling and then align short initial sequence fragments to reference genomes to decide whether to retain or reject a read. SquiggleNet [311] employs convolutional neural networks trained directly on raw nanopore electrical signals to classify molecules as target or non-target in real time, without requiring basecalling or full sequence alignment. Together, these tools provide flexible strategies for pathogen enrichment and targeted sequencing.
Despite its versatility, ONT adaptive sampling faces several limitations in transcriptomic applications. The relatively short length of mRNA molecules reduces the potential benefit of early read rejection, while delays in basecalling and alignment often mean that a substantial portion of each molecule is sequenced before a decision is made [304], [312]. Frequent rejection events can also decrease pore utilization efficiency and overall sequencing yield [303].
One of the most important advantages of nanopore sequencing is its capacity for real-time data generation [313]. Nanopore DRS enables rapid detection of RNA pathogens directly from clinical or field samples, with sequencing and analysis occurring concurrently. This capability has been demonstrated in studies where near-complete viral genomes and strain-level information were obtained within hours of sequencing, such as DRS of porcine reproductive and respiratory syndrome virus (PRRSV) from clinical samples, enabling accurate strain discrimination and detection of co-infections [314]. Real-time sequencing allows early identification of viral species and strains, often before a sequencing run is completed [315]. In addition, because DRS preserves native RNA molecules, it offers the unique potential to detect RNA modifications alongside sequence information, raising the possibility of monitoring epitranscriptomic signatures associated with virulence, host adaptation, or antiviral resistance [316]. As portable nanopore platforms continue to improve, DRS-based workflows are becoming increasingly feasible in decentralized or resource-limited settings. This opens new opportunities for on-site pathogen surveillance, rapid outbreak monitoring, and integration of transcriptomic and epitranscriptomic data into public health decision-making [317], [318], [319].
Bacteria, archaea and metatranscriptomes
DRS has so far been less widely applied to bacteria, archaea and metatranscriptomes, largely because commercial kits compatible with non-poly(A) RNA molecules are not available. With recent technological advances, however, this approach is emerging as a powerful approach for dissecting bacterial, archaeal, and community-level metatranscriptomes, enabling information to be retrieved across multiple layers of biological regulation (Figure 4).
In prokaryotes, one major application of DRS is the single-molecule, full-length characterization of primary transcript architectures. In Vibrio parahaemolyticus, enzymatic poly(A) tailing followed by DRS enabled comprehensive mapping of TSS, TTS, and operon structures, revealing that many internal and antisense TSSs actually arise from overlapping genes and quantifying highly complex combinations of transcriptional units within individual operons, thereby refining annotations derived from srRNA-seq [320]. Functionally, this strategy has been used to resolve antibiotic resistance repertoires in Klebsiella pneumoniae, simultaneously profiling the content of plasmid-borne resistance genes and their instantaneous expression levels; within ~10 h, expression signals for ≥ 35% of resistance genes can be detected and multiple co-transcribed operons composed of resistance determinants (for example, rmtB-blaTEM-1 and aac(6′)-Ib-cr-blaOXA-1-catB4) can be delineated [321]. In E. coli, native RNA reads uncovered extensive heterogeneity in TSSs, TTSs, 5′/3′ UTR and operon organization, and revealed a diversity of transcript isoforms far exceeding that reconstructed from short-read data [191], [209]. In the plant pathogen Dickeya dadantii, virulence-associated genes are frequently embedded within complex, condition-dependent transcriptional units whose composition and boundaries shift markedly in response to environmental cues [322]. In S. aureus, native RNA reads enabled construction of a “discontinuous operon atlas”, identifying numerous operons that are split, interlaced, or rearranged across distant loci, thereby challenging the traditional view of strictly continuous operons [323]. Comparative analyses across multiple bacterial and archaeal species further indicate that archaea commonly exhibit abundant leaderless mRNAs, atypical initiation and termination patterns, and irregular transcriptional units, features that are difficult to reconstruct from short reads [324].
DRS is also well suited for interrogating rRNA and tRNA modifications and their dynamic remodeling under environmental stress. Early work on 16S rRNA demonstrated that can distinguish canonical from modified nucleotides within native 16S molecules, providing a direct, signal-level readout of modification status [325]. A subsequent systematic analysis of E. coli ribosomes used DRS to resolve 17 distinct RNA modifications across 36 positions, enabling simultaneous detection of multiple modified sites in both the 30S and 50S subunits [262]. Antibiotic-challenge experiments further showed that ribosome-targeting drugs trigger pronounced loss and rewiring of modifications around the A- and P-site regions, which appear in DRS data as characteristic “modification-loss fingerprints” [326]. Heat shock and other perturbations induce broad remodeling of rRNA and tRNA modification landscapes in E. coli, and many mRNA modifications previously reported by antibody-enrichment approaches may in fact be substantially less prevalent than initially proposed [209], [327], [328].
Beyond rRNA, DRS has enabled single-molecule dissection of tRNA structure, processing and modification. Direct nanopore sequencing of individual full-length tRNAs has shown that even highly structured, heavily modified tRNAs can be read continuously from 5′ to 3′, with both sequence and modification information retained within the same molecule. This proof-of-concept established a technical foundation for functional studies, demonstrating that isoacceptor and isodecoder tRNAs can be distinguished on the basis of their molecule-specific modification signatures [36]. Given that archaeal rRNAs and tRNAs often harbor exceptionally dense and chemically diverse modifications, full-length native reads provide a unique opportunity to track how modification patterns mature over time and how they contribute to archaeal adaptation to extreme environments [329]. In Pseudomonas aeruginosa clone C, DRS captured condition-dependent transcriptional programs for virulence factors, resistance determinants and metabolic regulators within a single experiment [330]. In P. aeruginosa, tRNA hydroxylation has been identified as a key epitranscriptomic regulator of metabolic state and, consequently, pathogenicity; full-length, modification-preserving tRNA reads from DRS allow specific modification combinations to be directly linked to virulence phenotypes [331]. In E. coli, integration of NAD tagSeq II with DRS revealed pronounced growth-stage-dependent changes in NAD+ capping across the transcriptome, suggesting that this redox-linked 5′ modification acts as a molecular sensor of metabolic status and growth dynamics [332]. Likewise, RNA-mediated immune pathways can be directly monitored by DRS. For example, in an RNA-activated Cas12a3 defense system, Cas12a3 cleaves tRNA tails to execute antibacterial immunity, and the resulting cleavage and processing patterns are detectable [333]. Collectively, these studies illustrate how it connects RNA modifications and processing events to core functional phenotypes such as virulence, stress tolerance, and immunity in bacteria and archaea. At the community level, DRS has been applied to metatranscriptomic and clinical settings. The DEMINERS framework increases the throughput and accuracy of DRS data, enabling comparative transcriptome analyses of complex clinical specimens and improving both the sensitivity and resolution of pathogen detection and expression profiling [334]. In food safety, combining direct metatranscriptomics with multiplex RT-PCR amplicon sequencing allows simultaneous detection of viable pathogens within complex food matrices, with DRS providing direct evidence for pathogen presence and active expression of virulence genes [335]. In environmental studies, optimization of RNA extraction protocols for nanopore DRS has enabled soil metatranscriptome profiling from highly inhibitory matrices, facilitating characterization of active microbial consortia and their expressed functional pathways [336]. In marine ecosystems, DRS-based metatranscriptomics of planktonic crustacean communities has revealed seasonal shifts in community composition and functional gene expression, underscoring the potential of DRS for time-series monitoring of active microbiomes in situ [337]. At present, modification analyses in metatranscriptomic DRS are still largely confined to global trends or abundant rRNA/tRNA species, and precise mapping of low-abundance mRNA modification sites remains challenging.
Finally, standardized protocols for preparing native and unmodified bacterial RNA, together with matched IVT controls, have provided a robust experimental and analytical foundation for these studies [301]. Overall, the application of DRS in prokaryotic systems and their metatranscriptomes is rapidly evolving from purely structural transcriptomics towards integrated epitranscriptomic, functional, and ecological analyses.
Plant development and environmental responses
Plant growth, development, and environmental adaptation rely on tightly coordinated regulation across multiple layers of RNA biology, including transcript structure, alternative processing, polyadenylation, RNA modification, and the activity of non-coding RNAs [338], [339]. Historically, these layers have been investigated using distinct experimental strategies, mainly srRNA-seq for expression and splicing, 3′-end sequencing for APA, and antibody-based approaches for RNA modifications, resulting in fragmented datasets that lack molecular linkage [340]. Consequently, it has remained difficult to determine whether different RNA regulatory features co-occur on the same transcript molecules or act independently within a given developmental or stress context.
DRS provides a technical advance by enabling the simultaneous interrogation of multiple RNA features on native RNA molecules. In many plant species, this integrative capacity has begun to reveal coordinated RNA regulatory states that were previously inaccessible [52], [341]. A representative example comes from rice, where DRS-based analyses jointly profiled m6A and m5C modifications across multiple tissues [33]. 3389 to 14,499 modified transcripts in different tissues carrying both modifications were detected, indicating widespread co-occurrence of epitranscriptomic marks that had previously been mapped separately. Importantly, these modifications were not uniformly distributed across all transcript isoforms of a gene; instead, specific isoforms, often defined by alternative 3′ ends or terminal exon usage, exhibited preferential modification, suggesting a tight coupling between RNA processing and epitranscriptomic regulation. In A. thaliana, direct sequencing of native RNAs revealed that m6A-modified transcripts frequently display distinct poly(A) tail length compared with unmodified transcripts, particularly under stress conditions [52]. Beyond protein coding RNAs, DRS has extended multilayer analyses to lncRNAs. 1149 novel lncRNAs from A. thaliana and their m6A modification and poly(A) tail features has been simultaneously profiled, revealing reduced methylation levels relative to protein-coding RNAs, a positive role of m6A in lncRNA abundance, and a length-independent contribution of poly(A) tails to lncRNA stability [211]. These studies illustrate how DRS expands plant RNA research from parallel analyses of isolated features into an integrated view of coordinated RNA states. Nanopore DRS enables mechanistic insights into how plants fine-tune gene expression during development, forming the foundation for the problem-driven discoveries in plants.
Plants are continuously exposed to fluctuating environmental conditions and pathogen challenges, requiring rapid and reversible reprogramming of gene expression [342]. While transcriptional induction and repression under stress have been extensively characterized using srRNA-seq, accumulating evidence indicates that stress adaptation in plants relies heavily on post-transcriptional regulation [218], [342]. A major limitation of conventional approaches is that these regulatory layers are typically assayed separately, making it difficult to determine how they are coordinated on the same RNA molecules during stress responses.
DRS has begun to address this challenge by enabling integrated analyses of multiple RNA features in stressed plant tissues. In A. thaliana, DRS-based profiling under salt stress revealed widespread remodeling of both mRNA processing and RNA modification landscapes [52], [269]. These studies showed that stress-responsive genes frequently exhibit coordinated changes in APA, isoform usage, and m6A modification, rather than isolated alterations in any single layer. Importantly, m6A-modified transcripts displayed distinct stability and protein output profiles under stress, linking epitranscriptomic regulation directly to physiological outcomes [343]. Similar multilayer regulatory patterns have been observed in other crop species [344], [345]. In maize roots exposed to short-term salt stress, DRS revealed extensive stress-induced transcriptome reorganization, including 2223 differential expressed isoforms, reduced poly(A) tail length, and decreased m5C signals [346]. These findings highlighted that stress responses in crops are not limited to changes in gene expression levels but involve coordinated reconfiguration of transcript structures and chemical modifications, which may contribute to stress tolerance mechanisms. DRS has also provided new insights into the role of RNA regulation in biotic stress responses. Researches from A. thaliana and apple demonstrated that m6A modification is essential for resistance against pathogens, with stress-induced changes in RNA modification patterns affecting the expression and processing of defense-related transcripts [347], [348]. Notably, these studies linked RNA modification status to specific transcript isoforms and processing outcomes, suggesting that epitranscriptomic regulation fine-tunes immune responses at a resolution beyond gene-level expression.
TEs are a defining feature of plant genomes, often accounting for the majority of genomic content in crop species [349]. Beyond their well-established roles in genome evolution and structural variation, increasing evidence suggests that TEs actively shape transcriptome diversity and stress responsiveness [350], [351]. However, dissecting TE-derived transcriptional regulation has long been challenging, as srRNA-seq struggles to resolve TE-associated transcript isoforms due to sequence repetitiveness, ambiguous read mapping, and fragmented transcript reconstruction [202]. By directly capturing full-length native RNA molecules spanning gene-TE junctions, DRS has revealed how TEs are transcriptionally integrated into host gene architectures. A landmark example comes from the cotton genus (Gossypium), whose genomes are characterized by massive TE expansion and frequent polyploidization. Tian et al. demonstrated that TEs exert a widespread and systematic impact on post-transcriptional regulation across eight cotton species [352]. Specifically, TE-associated isoforms were found to be pervasive within a substantial proportion of expressed genes, harboring distinct TE-derived features, including alternative terminal exons and intragenic TE insertions that create new alternative splice sites. Notably, TE-driven turnover of splice sites and regulatory sequences may have contributed to regulatory divergence following polyploidization in cotton, with 5255 genes showing lineage-specific divergence at the splicing level. Complementary insights have been obtained in A. thaliana, where DRS demonstrated that intragenic TEs can act as regulatory modules that are co-transcribed with host genes, and that their epigenetic states influence RNA polymerase II elongation and the usage of APA signals embedded within TE sequences [276]. These findings provide a mechanistic framework for understanding how TE-derived regulatory elements are conditionally activated, particularly in plants, where a sessile lifestyle and constant exposure to environmental and biotic stresses impose strong selective pressure for stress-responsive regulatory plasticity [353].
Allele-specific regulation is a pervasive yet underexplored layer of gene control in plants, particularly in hybrids and polyploids where multiple parental or subgenomic copies coexist within the same nucleus. In these systems, phenotypic variation, heterosis, and environmental adaptation often arise not simply from differences in gene presence or absence, but from allelic divergence in expression level, RNA processing, and post-transcriptional regulation [354], [355]. However, resolving such allelic complexity has long been challenging, as srRNA-seq lacks sufficient haplotype context to confidently assign reads to specific alleles, especially in highly similar homoeologous regions. Beyond expression quantification, DRS has revealed that allelic divergence frequently extends to transcript structure and RNA processing. In allotetraploid B. napus, Li et al. uncovered extensive subgenome-specific AS and APA, with thousands of homoeologous gene pairs exhibiting asymmetric isoform usage across tissues [356]. Similar patterns have been observed in other polyploid systems, where one allele preferentially produces truncated or extended transcript isoforms, suggesting that post-transcriptional regulation contributes to functional divergence of subgenome following polyploidization [357].
Long-read analyses of panicle development in hybrid rice illustrate the power of full-length transcript profiling to resolve allele-dependent APA landscapes. Using Iso-seq, Wu et al. showed that ~80% of expressed genes harbor multiple poly(A) sites and that APA patterns differ between hybrids and parental lines, with shortened 3′ UTRs associated with increased expression and miRNA-mediated regulation during spikelet development [358]. While not based on DRS, these findings highlight how long-read approaches uncover heritable APA regulation that is largely inaccessible to short reads. Importantly, DRS could further extend such analyses by coupling accurate APA detection with allele-specific RNA modification profiling on the same native transcripts, opening new avenues for dissecting multilayer allelic regulation in plants. Recent nanopore DRS studies in human and mouse systems have already demonstrated the feasibility of resolving allele-specific m6A modification patterns at single-molecule resolution [359], providing a proof-of-principle for extending similar epitranscriptomic analyses to plant transcriptomes.
Beyond descriptive transcriptome analyses, DRS has increasingly been applied to resolve concrete molecular mechanisms underlying plant development, with flowering-time control in A. thaliana serving as a paradigmatic example. Flowering integrates environmental cues and endogenous signals through multilayered regulation involving transcription, chromatin state, RNA processing, and RNA modification. Many key regulators in this pathway, such as FLC, FCA, FPA, and antisense transcripts like COOLAIR, are controlled predominantly at the post-transcriptional level, rendering them particularly amenable to DRS-based interrogation [360], [361].
Early DRS studies in A. thaliana revealed extensive heterogeneity in RNA 3′ end formation and transcript termination among flowering-related genes, uncovering APA events that were previously obscured by srRNA-seq [52]. Subsequent work demonstrated that RNA-binding proteins such as FPA promote premature transcription termination at specific loci, including nucleotide-binding leucine-rich repeat (NLR) genes and flowering regulators, thereby reshaping transcript architecture and downstream gene activity [362]. Further, DRS has enabled direct interrogation of RNA modification in flowering control. In the seedlings and inflorescence tissues of A. thaliana, DRS-based analyses showed that m6A deposition is enriched near stop codons and 3′ UTRs of flowering regulators, and that perturbation of m6A writers or readers leads to delayed flowering phenotypes [363], [364]. Crucially, DRS revealed isoform-specific m6A patterns, linking alternative RNA processing directly to epitranscriptomic regulation. By tracing how RNA processing and modification converge on specific regulatory genes, DRS enables a transition from global transcriptome surveys to mechanism-driven insights.
Taken together, studies across plant development and environmental responses highlight important advances enabled by nanopore DRS, whereby transcriptome analysis is no longer confined to parallel measurements of isolated RNA features but can instead capture coordinated RNA states on individual native molecules. DRS has revealed how multiple regulatory layers are jointly remodeled during development, stress adaptation, and genome evolution. In this sense, DRS functions not merely as a profiling technology, but as an analytical framework that links RNA-level coordination to phenotype-relevant mechanisms in complex plant systems.
Animal models and organ development research
Understanding organ development requires a comprehensive dissection of transcriptional regulation across multiple aspects, including gene expression dynamics, isoform choice, RNA processing (AS and APA), and post-transcriptional modifications. The resolution of information from these regulatory layers is constrained by the underlying RNA sequencing technology. Traditional srRNA-seq has transformed developmental biology; however, it relies on indirect reconstruction of transcript structures and often fails to resolve complex isoforms, repetitive regions, and long-range coupling between exons and 3′ UTR. In addition, RT and PCR introduce biases that obscure native RNA features. Nanopore DRS provides an alternative transcriptomic approach to address many of these limitations [9], [35], [51], [56], [325].
Animal models are particularly well-suited for DRS-based analyses. Developmental staging in organisms such as C. elegans, zebrafish, and mice allows time-resolved mapping of isoform programs. Genetic perturbation and tissue-specific sampling support inference linking developmental phenotypes to AS or APA, with structural transcript changes directly observable in vivo using DRS. Moreover, in organs with high isoform complexity (brain, heart, immune organs), DRS complements single-cell and short-read atlases by providing full-length transcript context, including isoform-resolved regulatory features such as epitranscriptomic signatures and tail dynamics.
A landmark demonstration of DRS in developmental biology is the full-length DRS analysis across C. elegans developmental stages, which reveals extensive unannotated isoforms and unexpectedly high transcriptome complexity [201]. This work illustrates several key advantages of DRS in development studies. First, DRS identifies novel isoforms that are specific to embryonic, larval, or young adult stages. Second, full-length coupling of DRS links splicing patterns to 3′ ends for analysis of developmental regulation of UTRs. Third, refined transcript annotations enable isoform-level hypotheses in developmental genetics. Beyond development, DRS time-course studies in C. elegans aging provide a methodological template for longitudinal DRS analyses of developmental organ maturation, as such longitudinal designs reveal coordinated transcriptomic remodeling across isoform usage, tail dynamics, and epitranscriptomic regulation [365].
Embryogenesis involves rapid cell fate decisions and dynamic AS. Long-read sequencing studies of developmental stage transitions identify unannotated isoforms and stage-dependent splicing within full-length data [366]. Early zebrafish development also undergoes dramatic changes in polyadenylation and translation during the maternal-to-zygotic transition. Combining DRS with polysome or ribosome profiling directly links poly(A)/UTR features to translational output. DRS is particularly powerful for mapping isoform switching during organogenesis (heart tube formation, neurogenesis, hematopoiesis); defining stage-specific patterns of 3′ UTR remodeling associated with translational control; and characterizing RNA modifications during developmental transitions.
DRS is now widely applied in the study of mammalian tissues, including multi-organ surveys. An example integrates nanopore DRS to map tissue-specific RNA landscapes across mouse organs under metabolic perturbations, illustrating the breadth of in vivo DRS readouts at the levels of isoforms, expression, and RNA metabolism [367]. Similarly, a porcine multi-tissue study with fetal samples profiles transcript isoforms and the m6A landscape across tissues using DRS [368], demonstrating that DRS can scale to complex mammalian physiology and support comparisons between fetal and adult tissue programs [369]. For developmental neurobiology, DRS approaches are important because many regulatory RNAs in neurogenesis and synaptogenesis are not strictly polyadenylated or are expressed in isoforms with complex ends. Therefore, expanded DRS can provide a more comprehensive view of the regulatory RNA landscape during brain development [370]. Using DRS across multiple mammalian species and an avian outgroup, a recent study revealed that a small subset of evolutionarily conserved isoforms accounts for most gene expression, while the majority of isoforms are species‑specific and lowly expressed. The identification of conserved and isoform‑specific m6A deposition, along with widespread coordinate splicing, highlights the evolutionary importance of epitranscriptomic regulation in maintaining functional transcript diversity and buffering transcriptome variation [371].
Developmental regulation often relies on the process of transcript maturation rather than transcript abundance. DRS captures transcript maturation through long reads spanning splice junctions and PAS, with extensions to nascent or chromatin-associated RNA. K. Choquet et al. employed the DRS approach in 12 human lymphoblastoid cell lines (LCLs), and identified allele-specific splicing patterns that are regulated by genetic variants, such as splice-site SNPs and distal intronic variants [370]. Notably, HLA class I genes exhibited frequent allele-specific splicing orders that co-occurred with AS and APA events, illustrating the complexity of RNA maturation regulation in immune-related loci [370]. In addition, the same study described genetic regulation of nascent poly(A) tail lengths in chromatin-associated RNA (including large allele-specific differences for ERAP2), suggesting a connection between polyadenylation dynamics and transcript abundance [370]. In developmental immunology and organ maturation, such allele-dependent programs may tune transcript timing, isoform choice, and abundance in a lineage- and stage-specific manner.
Vertebrate embryo studies in zebrafish and Xenopus laevis show that DRS demonstrate stage-specific isoform switching and poly(A)-tail dynamics during embryogenesis, supporting a role for post-transcriptional regulation in major developmental transitions [114]. In combination with modification profiling [372], [373], these studies indicate that DRS provides a unified view of isoform structure, processing, and epitranscriptomic state during development. Cardiac systems present a practical challenge because mitochondrial RNAs account for a large fraction of polyadenylated RNA in cardiomyocytes, masking lower-abundance nuclear transcripts. Adaptive sampling has been combined with DRS to reduce mitochondrial RNA abundance in mouse heart tissue and hiPSC-derived cardiomyocytes [194], [302]. This methodological adjustment improves detection of low-abundance transcripts (e.g., Ccl7, Rbm6) and supports precise characterization of isoform expression and AS during cardiomyocyte differentiation [374], [375]. This integrated DRS-based approach shows that selectively shifting the molecules sequenced can reveal previously undetected developmental regulators and improve mechanistic inference in organ development. Nanopore DRS overcomes key limitations of srRNA-seq by providing long-read, native RNA measurements that resolve full-length transcript structures, poly(A) tail dynamics, and RNA modifications. Across model organisms and mammalian systems, DRS offers a broadly applicable framework for characterizing transcript maturation and post-transcriptional regulation during organ development.
Medicine and disease research
Human diseases increasingly reflect dysregulated RNA processing rather than simple changes in gene expression. Alterations in splicing, polyadenylation, RNA modifications, stability, and translation collectively shape pathology. Nanopore DRS enables simultaneous analysis of full-length transcript structure and epitranscriptomic features on single molecules.
AS drives transcriptomic diversity and is frequently dysregulated in disease, altering coding potential and RNA fate without changing gene expression. srRNA-seq often misses complex or low-abundance isoforms. DRS enabling integrated analysis of splicing architecture alongside multiple regulatory features on the same molecule [9], [207], [376], [377].
In biomedical contexts, DRS has proven useful for resolving disease-associated splicing complexity. For example, DRS identified 32 distinct full-length BRCA1 isoforms, many harboring multiple coordinated exon-skipping events, including co-occurrence of known pathogenic deletions such as Δ9-10 with Δ21 [378]. In inherited retinal dystrophies, DRS enabled direct detection and quantification of full-length aberrant transcripts arising from cryptic splice sites, pseudoexon inclusion, and complex combinations of splicing defects, including low-abundance isoforms that are difficult to resolve with srRNA-seq [379]. In rare genetic disorders, integration of nanopore DRS or cDNA sequencing with genomic haplotypes has enabled allele-resolved analysis, linking heterozygous splice variants to allele-specific isoform expression, as demonstrated in McArdle disease [380]. More broadly, DRS-based analyses have identified novel isoforms, differential transcript usage, and poly(A) tail variation as potential biomarkers in conditions such as sepsis [381].
Despite these advantages, several limitations remain. Nanopore sequencing error rates are higher than those of Illumina platforms, often necessitating customized bioinformatic pipelines and, in some cases, short-read-based error correction for precise splice junction and open reading frame annotation [197], [377], [382]. In addition, full-length DRS reads typically miss a short segment at the extreme 5′ end (approximately 6−12 nt), meaning that accurate TSS mapping may require complementary assays. Quantification of isoform abundance, while steadily improving, is still less mature than gene-level quantification from short-read data, and hybrid experimental designs are therefore commonly employed [381], [382], [383].
APA is a widespread but underappreciated regulator of gene expression. By altering poly(A) site usage, APA reshapes 3′ UTR length and regulatory elements, influencing RNA stability, localization, and translation, and is frequently dysregulated in disease. DRS is uniquely suited to study APA because it sequences native RNA molecules from the 3′ end through the full poly(A) tail, generating a characteristic homopolymer signal that enables single-molecule mapping of transcript termini and estimation of poly(A) tail length [35], [60], [226], [228], [384]. Genome-wide DRS provides reproducible poly(A) tail measurements across a broad dynamic range, from a few nucleotides to ~400 nt in human cells, offering quantitative insight into 3′-end regulation beyond short-read capabilities. Related methods, including TERA-seq and Nano3P-seq, profile both polyadenylated and non-polyadenylated RNAs while preserving native 3′ ends, showing high concordance with annotated APA sites. End-capture nanopore strategies further resolve RNA processing and decay intermediates at single-molecule resolution. By avoiding RT and PCR, DRS reduces protocol bias, especially valuable for variable-quality clinical samples.
At the biological level, nanopore DRS and related end-capture methods reveal extensive heterogeneity in poly(A) tail length and composition across transcript isoforms. Genome-wide analyses show broad, isoform-specific tail length distributions that correlate with RNA stability and decay during vertebrate embryogenesis [35], [384]. In human leukemia cells, DRS uncovered a negative correlation between poly(A) tail length and steady-state RNA abundance and stability, challenging the simplistic view that longer tails universally confer increased stability and instead revealing pathway-specific tail-length regimes [60]. Integrative analyses across multiple datasets further demonstrate that both tail length and tail composition, including the presence of non-adenosine residues, regulate mRNA stability and translation in a context-dependent manner, with short or heterogeneous tails often marking highly expressed and efficiently translated transcripts [226], [232], [234], [235], [236], [385].
Specialized nanopore methods such as Nano3P-seq and PolyTailor explicitly quantify poly(A) tail composition, enabling detection of non-A residues and mixed tails as additional regulatory layers [384], [386]. These features have functional consequences across diverse systems. In human cells, allele-specific DRS has identified genetic variants that modulate poly(A) tail length in concert with splicing order and APA choice, particularly in HLA class I genes, linking 3′-end regulation to inter-individual differences in immune gene expression [370].
Despite progress, standard nanopore DRS enriches for poly(A)+ RNAs and typically requires tails > 10 nt, potentially underrepresenting short- or tail-less transcripts. Modified approaches, such as poly(I) tailing or Nano3P-seq, improve coverage. Nevertheless, DRS enables profiling of native RNA termini and poly(A) tails, advancing understanding of APA and 3′-end regulation.
RNA chemical modifications, including m6A, Ψ, and Nm, dynamically regulate RNA structure, stability, splicing, and translation. Studying these effects requires detection in native transcript context. Nanopore DRS provides a direct, signal-based approach for isoform-resolved epitranscriptomic analysis. As native RNA passes through a nanopore, each k-mer produces a characteristic ionic current signature [34], [114]. Chemical modifications alter current intensity and dwell time, generating detectable deviations captured either as basecalling errors or through modeling of raw signal traces [34], [114], [186], [387].
A key advantage of DRS is preservation of full-length native RNA molecules, allowing modification sites to be mapped within precise transcript contexts, including specific splice isoforms, UTRs, and overlapping transcripts [26], [114], [118], [120], [186], [256]. By maintaining linkage between modification and transcript architecture, DRS overcomes limitations of short-read methods that fragment RNA and obscure isoform assignment [34], [118], [120], [240], [241], [277], [388]. The field is supported by growing resources and tools [26], [240]. Databases such as DirectRMDB compile DRS-derived modification maps across species, while computational methods including Nanocompore, xPore, CHEUI, TandemMod, and ModQuant detect differential and multi-type modifications, estimate stoichiometry, and assess co-occurrence on single molecules [34], [119], [177], [182], [185], [389], [390]. These advances enable mechanistic analysis of RNA-modifying enzyme perturbations underlying disease.
Collectively, by converting modification-induced signal changes into single-molecule calls on full-length RNAs, nanopore DRS overcomes several limitations of conventional epitranscriptomic methods, enabling isoform-resolved analysis of RNA modification pathways in health and disease.
Nanopore DRS enables integrated analysis of RNA regulation across multiple layers. AS, APA, and RNA modifications coordinately shaping RNA fate, yet srRNA-seq typically analyzes them separately and loses isoform-level linkage between processing events [200], [391], [392], [393], [394], [395]. Mechanistically, AS and APA are tightly coupled: 3′ end processing influence exon selection, while splicing decisions reshape 3′ UTR architecture and poly(A) site choice, together modulating coding potential, mRNA stability, localization, and translation [237], [393], [396], and thereby influencing processes such as apoptosis, immune responses, and oncogenic signaling [200], [217], [391], [392], [394], [395], [397], [398]. RNA modifications further interact with AS, APA, and RNA-binding proteins [399], [400], [401], [402], [403], [404]. Epitranscriptomic studies increasingly highlight that these modifications function within integrated regulatory networks rather than as isolated marks [400], [401], [402], [403], [404].
Recent work has highlighted long-read and third-generation sequencing technologies as essential for quantifying coupled AS-APA events and for resolving full-length transcript isoforms in which multiple processing decisions co-occur [217], [237], [394]. By preserving the physical linkage between splice patterns, 3′ UTR architecture, and, uniquely in the case of DRS, native RNA modification states on the same molecule, these approaches enable direct investigation of coordinated regulatory programs that are largely invisible to fragmented short-read data or mark-specific assays [200], [217], [237], [394], [402], [403]. Such integrated readouts reveal how multiple layers of RNA processing are orchestrated on individual transcripts.
Dysregulation of AS, APA, and RNA modification contributes to diverse diseases, including cancer, cardiovascular and neurodegenerative disorders, and immune dysfunction [200], [217], [391], [392], [393], [394], [397], [398], [399], [401], [404]. Increasingly, studies emphasize integrative analyses that combine isoform usage, RNA processing, epitranscriptomic states, and functional outputs to define disease-specific regulatory programs and therapeutic targets [200], [394], [399], [400], [402], [403]. Long-read DRS embodies this systems-level perspective, enabling isoform-resolved analysis of coordinated RNA regulatory layers that are often rewired in pathological states.
Beyond structure and modification, RNA stability and translational efficiency are key endpoints of RNA regulation, with altered decay or translation reshaping protein output in disease. Studying these processes requires isoform-specific resolution. DRS provides molecule-level readouts of RNA turnover, including transcript integrity, modification patterns, and direct measurement of poly(A) tail length [60], [120], [226], enabling isoform-resolved analysis of tail dynamics across genes and biological conditions.
Genome-wide DRS analyses reveal noncanonical relationships between poly(A) tail length, RNA abundance, and stability. In human leukemia cells, shorter-tailed transcripts were often more abundant and stable [60], challenging the traditional long tail equals stability model. Instead, RNA fate appears context-dependent, shaped by specific tail-length regimes and tail dynamics. Consistent with TAIL-seq and related studies, poly(A) tail length and 3′-end modifications influence both RNA decay and translational efficiency, directly linking 3′-end regulation to protein output [60], [226], [232].
DRS-based analytical frameworks, such as NanoTrans and DEMINERS, extend these insights by jointly quantifying transcript isoforms, poly(A) tail features, and RNA modification states, enabling comparative analyses across normal and disease conditions or in response to therapeutic perturbations [120], [206]. Application of epitranscriptomic callers to DRS data, including pum6a and RNANO, has further connected modification dynamics to altered RNA stability in cancer, exemplified by hypoxia-regulated m6A demethylase activity in gastric tumors [405]. By integrating transcript structure and regulatory features at single-molecule resolution, nanopore DRS reveals coordinated changes in RNA stability and translation that drive disease progression and therapeutic response.
In prostate cancer, Nm of rRNA is a key regulator of translation. EZH2 binds the Nm enzyme fibrillarin (FBL) and increases rRNA Nm, which enhances global and IRES‑dependent translation and supports cancer cell growth [406]. Besides mapping Nm sites in rRNA, nanopore DRS combined with the NanoNm machine‑learning tool showed that internal Nm on mRNA increases mRNA stability and expression, is linked to widespread 3′ UTR shortening, and thereby promotes prostate cancer progression [178], [291], [407]. Studies have focused on the regulatory role of METTL3-mediated m6A modification in AS [408]. Most recently, Yi et.al. revealed that the interplay between EZH2 and m6A regulatory pathways [409] using DRS.
For Breast cancer, in ER⁺ breast cancer models, Nanopore DRS has shown that METTL3-dependent changes in m6A sites can be linked to pathways associated with sensitivity or resistance to endocrine therapy, thereby providing site-level evidence for mechanisms of resistance [410]. Further work indicates that different molecular subtypes exhibit subtype-specific patterns of m6A hypomethylation or hypermethylation, and gene-engineering experiments have verified the DRACH motif-dependent activity of ALKBH5, suggesting that m6A helps shape tumor heterogeneity [411]. Beyond m6A, long-read approaches have also been applied to profile the m5C epitranscriptome of breast cancer mRNAs, and, together with gene editing, to explore its regulatory networks, thereby extending the use of DRS to multiple modification layers [412].
For Hematological malignancies, DRS is used to analyze the transcriptome and epitranscriptome of human leukemia cell lines [60]. In acute myeloid leukemia (AML), azacitidine (AZA) treatment induces remodeling of mRNA m5C and has led to the proposal of prognostic biomarkers [413]. After both AZA and venetoclax (VEN) therapy, a panoramic analysis of m6A alterations suggests that RNA modifications are both a mechanism and an indicator of therapeutic efficacy [414]. DRS has revealed a novel mechanism of immunotherapy resistance mediated by intron retention in the CD19 gene and selective splicing of the 5′ UTR in the CD20 gene [415], [416]. These findings highlight the ability of DRS to directly capture “structurally resistant transcripts”.
In non‑small cell lung cancer, studies have linked aberrant m6A patterns to prognostic genes for risk stratification [417]. DRS can also perform single-molecule methylation analysis of small RNAs in blood and identify lung cancer-associated methylation patterns with diagnostic potential [418]. In colorectal cancer, DRS has enabled simultaneous profiling of RNA modifications and alternative splicing, uncovering tumor‑associated exon‑skipping events with biomarker potential. Loss of exon ENSE00001632812 in the MYH11‑201 isoform was consistently observed in tumor tissues and validated in TCGA cohorts, supporting its diagnostic relevance. By integrating modification calling with splicing analysis, this approach provides a framework for identifying cis‑regulatory relationships between RNA modifications and aberrant splicing, highlighting the power of long‑read sequencing to dissect epitranscriptomic regulation in colorectal cancer [419]. In lung adenocarcinoma, CMTR2 mutations lead to splicing defects and expose therapeutic vulnerabilities, highlighting the value of long-read sequencing for dissecting splicing mechanisms [98].
Nanopore DRS can be used to resolve m6A modifications on noncoding RNAs, providing complementary validation alongside MeRIP-seq [420]. Studies also show that the modification landscape correlates with tumor grade [421]. In addition, the lncRNA CHROMR is associated with patient survival, offering clues for potential biomarkers [422]. Combined with CRISPRi functional screening, these lncRNA candidates can be further advanced toward actionable therapeutic targets [423].
Moreover, Nanopore sequencing detected ribosomal protein-associated m5C changes and linked them to oxidative stress, metabolic reprogramming, and immune responses [424]. In clear cell renal cell carcinoma, Nanopore sequencing has been used to integrate the m6A methylome with the transcriptome, and long-read RNA sequencing of archived tissues has enabled the discovery of novel transcripts [425], [426]. In hereditary leiomyomatosis and renal cell carcinoma, it can directly captured cryptic fumarate hydratase (FH) splicing mutations [427]. DRS has been used to systematically map the dynamic landscape of m6A and m5C modifications in muscle-invasive bladder cancer and to assess their prognostic value [428], [429]. Nanopore RNA assembly improves the completeness of the transcriptomic landscape and enables the discovery of novel functional transcripts [430]. Transcript-level features suggest value for stratifying aggressive subtypes [431]. Prmt5-deficient B cells exhibit increased RNA-processing complexity and are associated with slowed colorectal tumor progression, indicating that RNA processing in immune cells can also influence the tumor ecosystem [432]. DRS is demonstrating strong multidimensional analytical power in pan-cancer research. First, at the level of transcriptome architecture, it enables the construction of a single-gene transcript atlas [433], identification of oncogenic gene fusions [434], [435] and tumor-specific splicing variants [436]. Second, at the epitranscriptomic level, DRS can be used to calling multiple RNA modifications in rRNAs [437], and lncRNAs [438], [439], and reveal their biological significance, including m6A [440], m5C, Ψ [441], and A-to-I [441], [442]. Nanopore-based epitranscriptomic fingerprinting of rRNA Nm and related marks can distinguish normal tissues, tissue-of-origin, and tumor subtypes, revealing stable, cancer-specific rRNA modification signatures that may serve as diagnostic biomarkers and inform ribosome-targeted therapies [437], which can enable the discovery of novel cancer diagnostic biomarkers [443]. In addition, DRS can dynamically monitor post-transcriptional regulatory features such as poly(A) tail [444], [445].
Nanopore DRS can bridge the chain from variant to isoform and then to phenotype. In human muscle cells, it can directly capture DUX4-activated repeats and isoforms, thereby completing disease-relevant transcript annotations [446]; in congenital myopathies, it enables transcript-level validation of the pathogenicity of biallelic DST-b variants [447]; and for suspected splicing variants, allele-specific isoform analysis can be performed [380]. DRS can interpret the pathogenicity of BRCA1 exon duplications that generate fusion transcripts [448].
In amyotrophic lateral sclerosis/frontotemporal dementia (ALS/FTD), DRS directly revealed aberrant cryptic polyadenylation resulting from TDP-43 protein loss [449], which may be associated with METTL3 deficiency [450], and enabled the construction of an m6A modification atlas in motor neurons [451]. In Alzheimer’s disease, DRS allows precise discrimination of disease subtypes [189]. In addition, DRS has been used to evaluate the impact of environmental toxins on the epitranscriptome of brain organoids [452], and to elucidate a mechanism whereby m6A modification regulates neuronal ferroptosis after intracerebral hemorrhage via Vdac3 [453]. Researchers generated a full-length transcriptomic atlas of abdominal aortic aneurysm and identified novel transcripts [454]. They also revealed an emerging role of m6A RNA modification in cardiovascular diseases [455]. In heart failure, dysregulation of hnRNPL affects AS [456]. In patients with sepsis, blood-based DRS can be used to identify both co-transcriptional and post-transcriptional disease biomarkers [381]. Studies have reported sex-specific transcriptomic and epigenomic characteristics in fear learning similar to those observed in post-traumatic stress disorder [457].
Framing RNA regulation by molecular mechanisms rather than disease categories underscores DRS as a versatile platform. Core processes, splicing, polyadenylation, RNA modification, and stability, are recurrently disrupted across diseases, and DRS enables systematic, isoform-resolved analysis in clinical samples, supporting cross-disease comparisons and shared regulatory insights. DRS is positioned to become a key tool for dissecting RNA regulatory mechanisms and advancing precision medicine.
Clinical and translational applications: RNA vaccines, RNA drugs, and diagnostic markers
The clinical relevance of nanopore DRS derives from its ability to capture multiple layers of RNA regulation with disease phenotypes [63]. By providing direct access to the epitranscriptome, DRS enables mechanistic insights into gene regulation and RNA function that are increasingly important for clinical diagnostics and therapeutic development [26]. In contrast to srRNA-seq, which fragments transcripts and relies on computational reconstruction, DRS identifies disease‑specific epitranscriptomic signatures and pathogenic isoforms without PCR amplification [458], thereby reducing technical bias and improving biological interpretability [189].
These technical advantages position DRS as a powerful platform for RNA biomarker discovery across diverse disease contexts. In cancer biology, the RNA modification landscape is tightly linked to tumor initiation and progression, with modifications such as m6A modulating oncogenic signaling pathways and tumor immune evasion [459]. Beyond cancer, recent DRS studies in sepsis have identified co‑ and post‑transcriptional biomarkers, including poly(A) tail length variation, RNA modification dynamics, and isoform usage patterns, providing deeper insights into disease states than traditional sequencing approaches [460]. Similarly, DRS has revealed multilayered epitranscriptomic remodeling in macrophages following M. tuberculosis infection, underscoring its diagnostic potential in infectious diseases [62]. Conventional RNA profiling methods are often limited by amplification bias and loss of native RNA features, whereas DRS offers a long‑read, amplification‑free alternative that preserves native RNA modifications and molecular heterogeneity, further expanding its utility for biomarker discovery and clinical applications [9], [461], [462].
Likewise, nanopore DRS enables comprehensive profiling of viral transcriptomes, such as SARS‑CoV‑2, revealing complex RNA isoforms and modification states that inform viral replication strategies and pathogenic mechanisms [279], [334]. More broadly, DRS is emerging as a transformative tool in clinical diagnostics by enabling direct detection of RNA modifications and post‑transcriptional regulatory features that serve as sensitive biomarkers across a wide range of diseases [442], [459], [463]. Continued advances in modification‑calling accuracy and stoichiometry estimation further enhance biomarker specificity and interpretability [381], particularly as disease associated alterations in RNA modification patterns often precede detectable changes in transcript abundance.
Beyond diagnostics, nanopore DRS has emerged as an enabling technology for RNA‑based therapeutics, particularly in the rapidly expanding fields of mRNA vaccines and immunotherapy. mRNA‑based strategies hold broad potential for infectious diseases, cancer vaccines, and personalized immunotherapies, in which patient‑specific neoantigens can be encoded into mRNA constructs to elicit tailored antitumor immune responses [464], [465], [466]. The relatively low production costs and manufacturing simplicity of mRNA therapeutics further support their potential for globally accessible treatment strategies [467]. However, the rapid clinical deployment of mRNA vaccines has created a critical need for high‑resolution technologies capable of assessing RNA quality, structural integrity, and modification states. DRS has proven particularly effective in this context by enabling direct, full‑length characterization of therapeutic RNA molecules, thereby supporting quality control, optimization, and regulatory evaluation of mRNA‑based therapeutics [468].
DRS enables direct evaluation of vaccine RNA by identifying truncated transcripts, poly(A) tail length heterogeneity, and synthetic nucleotide modifications, such as Ψ and m6A, that critically influence translation efficiency and immunogenicity [468]. These analytical capabilities underpin emerging frameworks for comprehensive mRNA vaccine quality control and batch‑to‑batch consistency testing [467], [468]. Moreover, the real‑time nature of nanopore sequencing facilitates longitudinal monitoring of RNA integrity and modification drift during storage, transportation, and deployment, representing a key capability for ensuring the reliability of global mRNA vaccine distribution pipelines [467].
In parallel with vaccine applications, a broad range of RNA‑based therapeutics, including mRNA drugs, antisense oligonucleotides (ASOs), siRNAs, and RNA‑editing constructs, require precise characterization of RNA stability, structure, and modification states, all of which can be directly assessed by nanopore DRS [460]. Among these features, m6A modifications play central roles in regulating cancer progression, immune homeostasis, metabolic disorders, neurocognitive function, and stem cell differentiation, underscoring their relevance for therapeutic RNA design and optimization [469], [470], [471], [472]. Beyond m6A, additional RNA modifications, including m7G, m5C, m1A, Ψ, and Nm, have been implicated in diverse disease pathways and represent promising targets for next‑generation RNA therapeutics [473], [474].
The development of mRNA‑encoded therapeutic antibodies by companies such as BioNTech and Moderna further illustrates the clinical potential of RNA‑based technologies, with multiple candidates currently in preclinical and clinical development targeting infectious diseases, cancer, and toxic exposures [475], [476], [477]. In this context, DRS provides a multidimensional view of RNA therapeutic performance and quality [478]. Beyond mRNA, modifications in mitochondrial RNA, tRNA, and diverse noncoding RNAs have been increasingly implicated in disease pathogenesis [271], [474], and ongoing advances in DRS continue to extend its applicability to these clinically relevant RNA classes.
Despite its transformative potential, DRS faces several challenges that must be addressed before widespread clinical adoption. Sequencing throughput remains lower than that of short‑read Illumina platforms, and RNA input requirements are comparatively high. Limited RNA yields from clinical specimens, particularly liquid biopsies, further constrain applicability [479], although sample multiplexing strategies may partially mitigate this limitation [37], [334]. In addition, short RNA molecules are inefficiently captured by nanopores; however, optimization of acquisition parameters within ONT’s MinKNOW software has been shown to substantially improve short‑RNA recovery [192]. DRS also exhibits systematic signal loss of approximately 15 nts at read termini, which can affect basecalling accuracy and transcript boundary resolution [480]. Although recent advances, including ONT’s SQK-RNA004 chemistry and updated flow cell designs, have improved sequencing throughput and enabled more reliable direct detection of RNA modifications such as m6A and Ψ, challenges related to RNA chemical stability and the lack of standardized regulatory and analytical frameworks continue to limit clinical implementation. These limitations are particularly relevant for the evaluation and quality control of mRNA vaccines and RNA‑based therapeutics [63].
Native RNA modification profiles offer distinct advantages over conventional expression‑based biomarkers for early disease detection and therapeutic response monitoring. Accumulating evidence indicates that epitranscriptomic alterations often precede measurable changes in transcript abundance, positioning RNA modifications as more sensitive and dynamic indicators of disease state [459]. Recent work in sepsis highlights this potential, demonstrating that nanopore DRS can directly sequence native RNA from minimally processed clinical samples, thereby enabling a viable path toward real‑time molecular diagnosis [381]. In parallel, by directly interrogating RNA structure, integrity, and modification authenticity, DRS complements established analytical techniques such as high‑performance liquid chromatography and mass spectrometry, providing a more comprehensive assessment of therapeutic RNA products. In summary, by delivering an unprecedented view of the transcriptome and epitranscriptome in their native state, nanopore DRS offers clear advantages for mRNA vaccine quality control, RNA biomarker discovery, and therapeutic RNA monitoring. Continued advances in AI‑powered basecalling, nanopore signal modeling, pore chemistry optimization, and standardized benchmarking frameworks are expected to further enhance accuracy, reproducibility, and interpretability. Together, these developments are likely to substantially expand the clinical reach of nanopore DRS, ultimately positioning it as an important technology for precision medicine applications.