Outlook: toward integrative, haplotype-aware RNA biology
From feature catalogues to multilayer regulatory logic
DRS has rapidly moved beyond proof-of-concept demonstrations toward integrative discovery, fulfilling a key early promise of the technology: the ability to read transcript structure, poly(A) properties, and multiple RNA modification signals simultaneously on native RNA molecules [51], [120]. This integrated readout can reduce experimental fragmentation compared with running parallel assays for isoforms, 3’ ends, and epitranscriptomics, although the maturity and reliability of these individual readouts are not yet equivalent across applications [202]. Accordingly, DRS is increasingly being considered not only as an alternative to conventional RNA-seq, but also as a potentially useful framework for studying multilayer RNA regulation in plant transcriptomics. A practical application of this transition is timely as the output of DRS is unusually model-ready. Each read naturally yields structured attributes, isoform identity, cleavage site choice, tail length estimates, and modification probabilities, that can be assembled into transcript- or gene-level knowledge graphs. Such graphs may provide a useful substrate for representation learning and, in some settings, may be compatible with large language model (LLM)-assisted biological reasoning, as has begun to be explored in crop-focused knowledge graph frameworks [38], [687]. Standardized DRS-derived feature sets could therefore serve as one possible bridge between experimental transcriptomics and AI-assisted hypothesis generation, although the biological validity and practical utility of such frameworks will require careful benchmarking (Figure 10A).
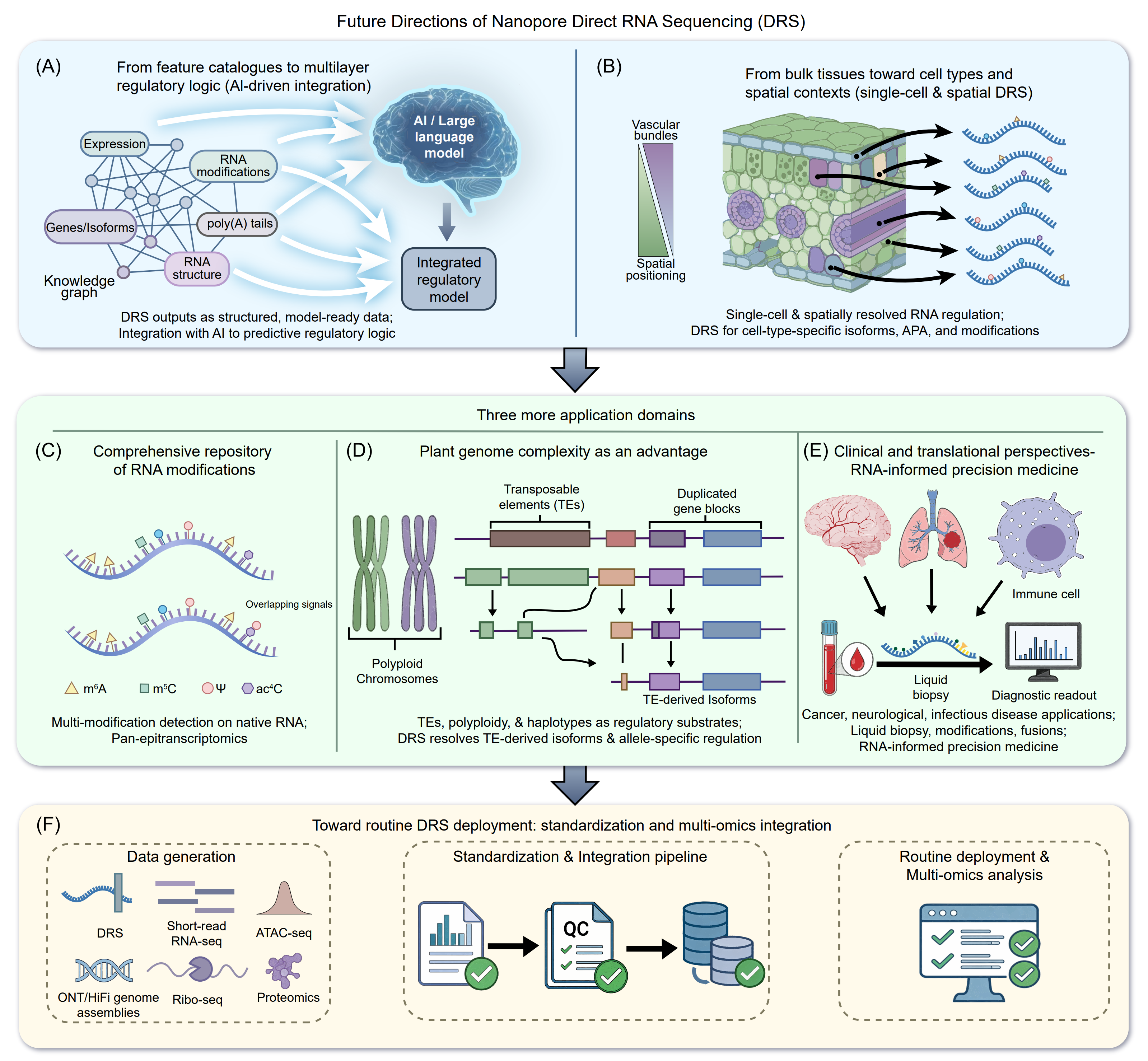
Figure 10. Outlook of nanopore DRS. (A) From feature catalogues to multilayer regulatory logic: DRS integrates isoforms, poly(A) tails, and RNA modifications into structured data frameworks compatible with AI and knowledge-graph models. (B) From bulk to single-cell and spatial contexts: emerging strategies aim to resolve cell-type-specific RNA regulation. (C) Expanding epitranscriptomics: multi-modification detection beyond single marks enables integrative RNA state analysis. (D) Genome complexity as opportunity: DRS resolves TE-derived isoforms and haplotype-specific regulation. (E) Clinical translation: workflow harmonization and multi-omics integration support scalable research and RNA-informed precision medicine. (F) Standardization and multi-omics integration form the foundation for routine and scalable DRS deployment.
From bulk tissues toward cell types and spatial contexts
Bulk DRS studies consistently imply that RNA regulation is highly context-specific, particularly for isoform switching, APA remodeling, and RNA modification dynamics. However, most current mechanistic inference still relies on tissue averages. Early long-read single-nucleus approaches in plants, although based on cDNA rather than native RNA, have already shown that AS and APA patterns differ sharply across cell populations, underscoring the value of long reads for resolving regulatory heterogeneity [688]. True single-cell DRS remains constrained by input requirements, signal noise, and cost, particularly when modification inference is included (Figure 10B) [54], [120]. In the near term, a more realistic path may be hybrid strategies: use short-read single-cell or single-nucleus data to define cell states and candidate regulatory programs, followed bulk or targeted DRS in enriched tissues to characterize multilayer RNA features with gather confidence [689], [690]. Even without native single-cell DRS, integrating bulk DRS with spatial transcriptomics or cell-type marker frameworks may help associate regulatory programs with anatomical contexts, although such assignments will often remain indirect [691], [692].
Accessing to a comprehensive repository of RNA modifications
Epitranscriptomics has so far been dominated by m6A and m5C, yet accumulating evidence indicates that additional modifications, such as hm5C and ac4C, contribute to development, stress responses, and long-distance RNA transport [693], [694]. DRS is particularly attractive in this context because it may, in principle, detect multiple modification signatures on the same native RNA molecules and relate them to isoform usage, APA decisions, and stability-associated features. A key future direction is to move from single-accession snapshots toward pan-epitranscriptomes, mirroring the conceptual shift introduced by pangenomes [202]. If sufficiently robust and transferable analytical pipelines become available, population-scale DRS could help distinguish conserved modification sites that form regulatory backbones from variable, genotype- or environment-specific marks that may tune phenotypes (Figure 10C). Methodologically, this expansion depends on robust and transferable modification callers. Recent benchmarking and transfer-learning studies suggest that nanopore signal perturbations induced by RNA modifications are sufficiently conserved to support cross-species model adaptation [175], [177], [180]. However, whether this will generalize across modification classes, sequence contexts, and chemistries remains to be established. These developments therefore point toward, rather than yet deliver, a broader repository of RNA modifications across species. In particular, low-abundance modifications and condition-specific marks are likely to remain difficult to catalogue systematically until both signal sensitivity and orthogonal reference datasets improve substantially.
Plant genome complexity as an advantage: TEs, haplotypes, and evolution
Plants are enriched for the two genome features that most challenge transcriptomics, TEs and polyploidy, but these same features make DRS particularly powerful. Long-read DRS can directly resolve TE-derived isoforms, chimeric gene-TE transcripts, and alternative termination within repetitive contexts that confound short reads [276], [352]. These studies reveal TE-derived transcripts are not annotation artifacts but regulatory entities shaped by RNAPII elongation, epigenetic state, and alternative poly(A) signal usage. In polyploid crops and diverse germplasm panels, DRS may also provide an allele- and haplotype-aware readout, supporting analysis of ASE and, potentially, allele-specific RNA modification and APA [202], [359]. If these applications become more robust, they could enable an evolutionary transcriptomics framework in which TE-derived isoforms and allele-specific RNA regulation are treated not merely as analytical noise, but also as possible substrates for adaptation (Figure 10D). Because polyploidization is pervasive across the plant kingdom, particularly in wild and non-model species [695], DRS may be especially useful for studying transcriptomic regulation in complex genomes where short-read approaches are often limited.
Clinical and Translational Perspectives: Toward RNA-Informed Precision Medicine
DRS offers a powerful framework for future clinical diagnostics by enabling direct, amplification-free profiling of full-length native RNAs together with their modification states (Figure 10E). However, its translational relevance remains contingent on overcoming key challenges, including analytical sensitivity, clinical-grade accuracy, reproducibility, and standardized workflows compatible with regulatory requirements [54], [189]. If these barriers can be addressed, DRS could contribute to a gradual shift in some settings from static, gene-centric measurements toward more dynamic, RNA-centered molecular profiling [696], [697].
In oncology and liquid biopsy applications, DRS may eventually help resolve oncogenic fusion transcripts, tumor-specific splice isoforms, and possibly aspects of RNA modification variation from limited circulating RNA inputs, although these use cases still require substantial validation and sensitivity improvements [698], [699]. In neurological and infectious diseases, DRS also offers a potential route for direct interrogation of pathogen and host transcriptomes, including RNA processing changes and candidate modification dynamics that may prove informative for diagnosis, disease monitoring, or therapeutic decision-making [700], [701]. Beyond diagnostics, DRS is being explored as a possible platform for quality control of RNA therapeutics, where single-molecule assessment of sequence integrity, poly(A) tail length, and engineered nucleotide content may be valuable for mRNA vaccines and RNA drugs. Taken together, these examples suggest translational potential, but routine clinical implementation will require rigorous analytical validation, standardized reporting, and demonstration of added value over existing assays.
Toward routine deployment: standardization and multi-omics integration
Despite its potential, the widespread adoption of DRS remains constrained by cost, signal noise, and analytical complexity, particularly for modification inference [51]. A realistic path forward may be task-specific standardization, with best-practice workflows for transcript structure and APA analysis, poly(A) tail profiling, and RNA modification calling, each accompanied by explicit quality-control metrics and computational expectations (Figure 10F). Community benchmarks and shared reference datasets will be essential for ensuring reproducibility across laboratories and application domains. In parallel, DRS may have the greatest impact when integrated with complementary omics rather than standalone use. Positioning DRS as the native-RNA layer alongside srRNA-seq, high-quality genome assemblies, chromatin accessibility assays, ribosome profiling, proteomics, and metabolomics has helped explain why transcript abundance alone does not always predict protein output and how post-transcriptional regulation reshapes biological responses [346], [352], [702]. In biomedical contexts, similar integrative strategies may support emerging applications in liquid biopsy [418], infectious disease surveillance [703], and RNA therapeutic quality control [704], especially where preserving the native linkage between sequence, processing state, and modification is advantageous for interpretation. Realizing the broader value of DRS will require continued advances in protocol optimization, computational efficiency, benchmarking, and regulatory-grade validation. As these barriers are progressively addressed, DRS may evolve from a powerful research technology into an increasingly important platform for mapping RNA regulation with greater molecular resolution. Another promising direction is the implementation of simultaneous DNA/RNA sequencing in a unified workflow, enabling the profiling of genomic variation, epigenetic features, transcript isoforms, poly(A) tails and RNA modifications from matched samples with reduced batch effects. Such integrated native-molecule sequencing could provide a more direct framework to connect genotype, chromatin and DNA modification status, transcript processing, and post-transcriptional regulation within the same biological context.