Application-oriented integration of nanopore DRS with multi-omics
A comprehensive understanding of gene regulation across various conditions requires integration across multiple molecular layers, spanning genetics, chromatin accessibility, transcription, RNA processing, RNA modification, translation, metabolism, and protein abundance. While genomics and transcriptomics provide information on genetic potential and RNA expression, they often fail to capture the post-transcriptional and post-translational regulatory processes that ultimately determine cellular phenotype. DRS occupies a unique position within this landscape by enabling native, full-length interrogation of RNA molecules, thereby serving as a critical bridge between upstream regulatory inputs and downstream functional outputs. Integrating DRS with other omics modalities offers a powerful framework for constructing coherent, mechanistic models of cellular regulation.
Genome-transcriptome integration and regulatory input-output mapping
One of the earliest demonstrations of DRS-enabled multi-omics integration was at the genome-transcriptome interface. By combining ONT DRS with long-read DNA sequencing, it became possible to simultaneously resolve genome architecture and native transcriptome regulation within a single analytical framework (Figure 9A). In yeast, the integration of DRS with PacBio long-read genome assembly enabled concurrent decoding of genomic structure, differential gene expression, DNA methylation, and RNA modification landscapes, illustrating how genetic variation and chromatin-level features are translated into mature RNA outputs [660]. This study established a conceptual blueprint for genome-to-transcriptome mapping in which DRS provides the missing link between DNA sequence and RNA regulatory state.
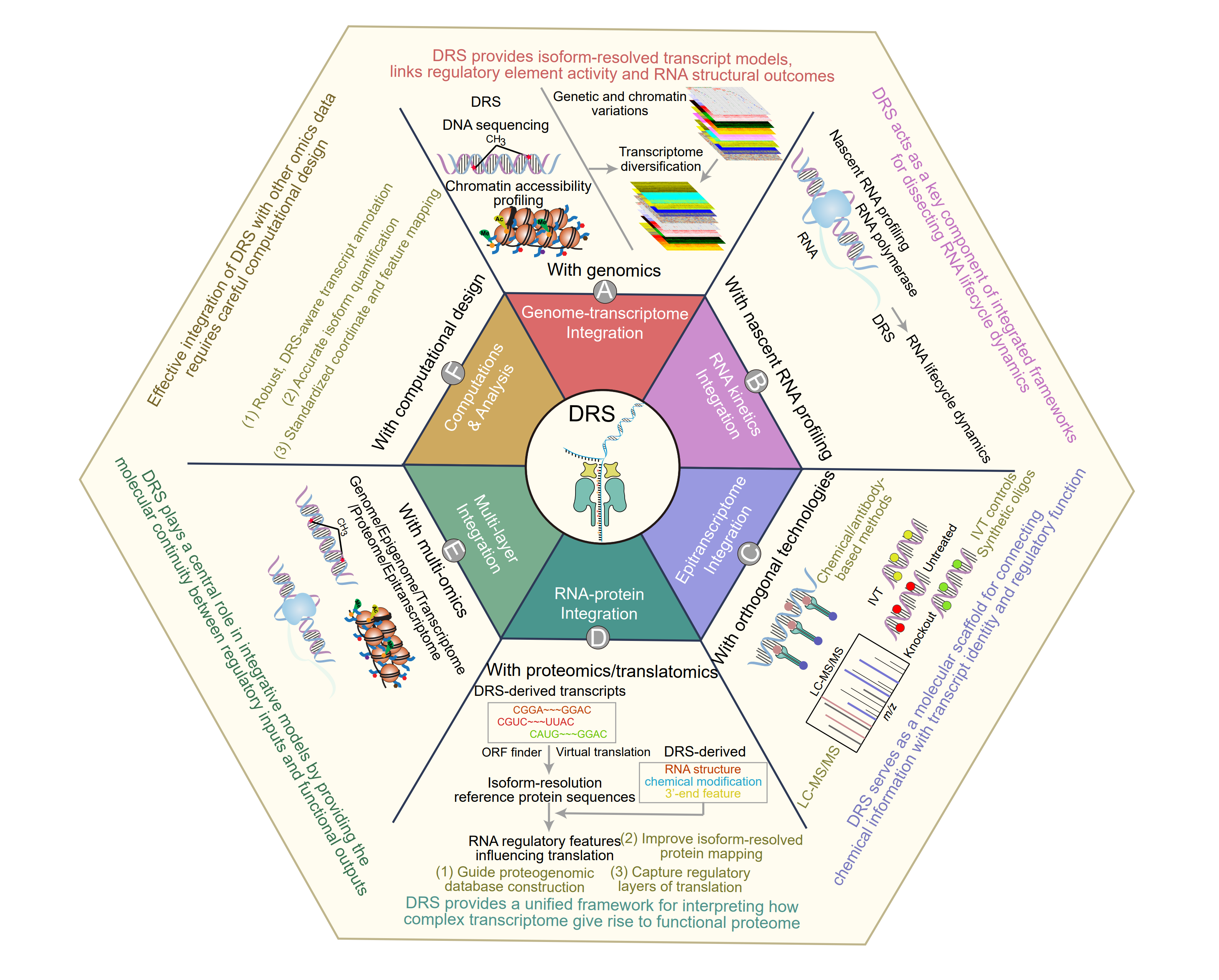
Figure 9. Nanopore direct RNA sequencing as a central hub for multi-omics integration. (A) In the integration with genomics, DRS provides isoform-resolved transcript models, links regulatory element activity and RNA structural outcomes. (B) In the integration with nascent RNA profiling, DRS acts as a key component of integrated frameworks for dissecting RNA lifecycle dynamics. (C) In the integration with orthogonal technologies for RNA chemical modifications, DRS serves as a molecular scaffold for connecting chemical information with transcript identity and regulatory function. (D) In the integration with proteomics, DRS provides a unified framework for interpreting how complex transcriptome give rise to functional proteome. (E) In the integration with multi-omics, DRS plays a central role in integrative models by providing the molecular continuity between regulatory inputs and functional outputs. (F) Effective integration of DRS with other omics data requires careful computational design.
Building on this foundation, more recent multi-omics strategies have integrated DRS with chromatin accessibility profiling and srRNA-seq to interrogate transcriptional regulation in complex mammalian systems [367]. By combining assay for transposase-accessible chromatin using sequencing (ATAC-seq) with native full-length RNA sequencing, these approaches systematically delineated tissue-specific RNA landscapes across mouse organs, uncovering thousands of previously unannotated transcripts and organ-specific isoforms. Importantly, these studies revealed that chromatin accessibility patterns alone are insufficient to predict mature transcriptomes, as extensive transcriptome diversification arises from downstream RNA processing. DRS was essential in this context, providing isoform-resolved transcript models that could not be reconstructed from srRNA-seq data and enabling direct linkage between regulatory element activity and RNA structural outcomes.
Integration with RNA lifecycle kinetics and nascent RNA profiling
Beyond static transcriptome annotation, DRS has become a key component of integrated frameworks for dissecting RNA lifecycle dynamics. RNA processing, stability, and decay are inherently temporal processes, yet conventional transcriptomic methods capture only steady-state snapshots. By coupling DRS with metabolic RNA labeling and machine learning-based signal analysis (Figure 9B), recent studies have enabled direct identification of newly synthesized RNA molecules and quantitative assessment of nascent RNA processing at isoform resolution [197], [370], [661], [662], [663]. These integrated approaches demonstrated that DRS can distinguish newly transcribed RNAs from pre-existing molecules based on characteristic signal features, allowing direct measurement of splicing kinetics, RNA maturation rates, and stability determinants without indirect inference. In particular, the ability of DRS to simultaneously capture transcript structure and poly(A) tail length has proven critical for understanding how isoform-specific 3′-end features influence RNA lifespan across cellular conditions.
Further expansion of this kinetic framework has been achieved through integration of DRS with complementary time-resolved sequencing strategies such as TimeLapse-seq [664]. By combining DRS with chemically encoded temporal information, these multi-compartment approaches provide a systems-level view of RNA flow across nuclear and cytoplasmic compartments. Such analyses reveal coordinated regulation of RNA processing, export, and decay, highlighting how transcript architecture and epitranscriptomic features jointly shape RNA fate in space and time.
Epitranscriptomic and chemical-layer integration
DRS-based integration has increasingly extended into the epitranscriptomic domain, where RNA chemical modifications introduce an additional and highly dynamic layer of post-transcriptional regulation. When used alone, nanopore DRS robustly detects modification-associated perturbations in ionic current signals on native RNA molecules, enabling transcriptome-wide discovery of candidate modification sites. However, signal-based detection by itself is inherently limited in chemical specificity and in controlling false positive arising from sequence context, RNA structure, or basecalling noise.
These limitations are effectively addressed through integration of DRS with orthogonal biochemical, genetic, and mass-spectrometric approaches, yielding comprehensive, isoform-resolved, and chemically defined epitranscriptomic maps (Figure 9C). In particular, coupling DRS with chemical- or antibody-based NGS methods, such as MeRIP-seq, miCLIP, bisulfite sequencing, or ac4C-seq, enables validation of candidate modification sites and assignment of modification classes at single-nucleotide resolution [33], [116], [387]. These approaches provide critical base-level specificity that complements the long-range contextual information captured by DRS. Mass spectrometry-based methods, including LC-MS/MS, further strengthen epitranscriptomic integration by directly measuring the chemical identity and global abundance of RNA modifications. Such measurements serve as gold-standard validation for DRS-inferred modification sites and regulatory circuits [256], [631], particularly for heavily modified RNA species such as tRNAs, where defined modification patterns (for example, within the T loop) can be independently confirmed. Together, DRS and mass spectrometry bridge molecular-scale chemical precision with transcriptome-scale structural context.
Genetic and experimental controls are equally essential for robust integration. IVT unmodified RNAs, as well as writer knockouts or knockdowns, provide critical negative controls that distinguish true modification signals from sequence- or context-dependent artifacts and enable calibration of detection thresholds [162], [177], [246], [256], [387]. These strategies are particularly important for training and benchmarking modification-calling algorithms, where false positives can otherwise propagate systematically. More recently, synthetic oligonucleotides and engineered epitranscriptomes containing defined combinations of RNA modifications have emerged as valuable sources of ground-truth training data. Such resources have enabled the development of multi-modification detection frameworks, including TandemMod and updated Dorado models, improving the accuracy of simultaneous detection of multiple modification types from single DRS datasets [177], [189]. These advances highlight the importance of combining experimental design with algorithmic innovation in epitranscriptomic analysis. A key advantage of DRS-based integration lies in its ability to preserve transcript-level context. By maintaining full-length RNA information, DRS allows chemical modification states to be associated with specific transcript isoforms, processing states, and expression levels, relationships that are largely inaccessible to short-read or site-centric approaches. This capability enables direct interrogation of how epitranscriptomic modifications intersect with AS, APA, RNA stability, and translation.
Collectively, these integrated strategies exemplify how DRS functions as a molecular scaffold that connects chemical information with transcript identity and regulatory function. Rather than replacing established epitranscriptomic methods, DRS complements them by embedding chemical modification data within data transcript architectures, thereby enabling a mechanistically grounded and systems-level understanding of RNA regulation.
RNA-to-protein integration through DRS: from proteogenomic databases to translational regulation
Bridging transcriptomic complexity with proteomic output remains a central challenge in functional genomics and life science research. Although high-throughput proteomics has matured rapidly, its interpretability is fundamentally constrained by the quality of transcript-derived protein sequence databases and by incomplete resolution of transcript isoform diversity. DRS offers a transformative framework for RNA-to-protein integration by providing native, full-length transcript information that directly informs proteogenomic database construction, isoform-resolved protein mapping, and mechanistic dissection of translational regulation (Figure 9D).
Accurate proteogenomic analysis depends critically on the completeness and correctness of the protein sequence database used for mass spectrometry searches. Conventional proteogenomic pipelines typically rely on reference transcript annotations or srRNA-seq-derived assemblies, which often fail to capture condition-specific isoforms, truncated transcripts, alternative reading frames, or noncanonical translation events. As a result, many peptides detected in proteomic experiments remain unmapped or are ambiguously assigned. DRS fundamentally improves proteogenomic database construction by directly defining full-length transcript isoforms without reliance on computational assembly. In Arabidopsis, incorporation of transcript isoforms derived from PacBio Iso-seq and ONT DRS into proteogenomic search databases substantially increased proteoform identification in bottom-up mass spectrometry compared with databases built from curated transcript annotations alone [665], underscoring the added value of DRS-resolved transcript diversity for proteomic analyses. Importantly, accumulating evidence from eukaryotic, viral, and bacterial systems indicates that protein search databases derived from DRS data can explicitly encode alternative translation initiation sites [52], [208], [666], premature termination events [667], [668], intron-retention-derived coding sequences [97], [668], and previously unannotated [666], [669], [670], including small, open reading frames, many of which are increasingly supported by proteomic evidence of active translation. By anchoring proteogenomic searches to native transcript structures, DRS increases peptide identification rates and reduces false-negative discoveries, thereby enhancing the depth and accuracy of proteomic analyses.
Beyond database construction, DRS enables more precise mapping of detected peptides to their transcript of origin, addressing a major limitation of traditional RNA-protein integration. Many protein-coding genes produce multiple transcript isoforms that encode highly similar or partially overlapping protein products. Short-read transcriptomics often cannot distinguish these isoforms reliably, leading to ambiguity in assigning peptides to specific transcript variants. DRS resolves this ambiguity by providing isoform-resolved transcript definitions that can be directly linked to protein sequences [81], [86], [90], [210]. When integrated with mass spectrometry data, this enables isoform-level protein inference, allowing peptides to be mapped to specific transcript variants rather than collapsed at the gene level [208], [665], [671]. Isoform-complete transcript definitions can be directly translated into protein sequences, enabling shared peptides to be correctly assigned to the expressed isoform while facilitating discovery and targeted analysis of isoform-unique peptides [210], [665], [671]. This approach directly resolves the ambiguity arising from genes that produce highly similar and overlapping protein products. Such resolution is critical for understanding functional diversification, as isoform-specific differences can affect protein domains, interaction motifs, subcellular localization signals, or post-translational modification sites.
A further strength of DRS-based RNA-to-protein integration lies in its ability to capture RNA regulatory features that directly influence translation but are largely invisible to conventional transcriptomic approaches. Translation is not determined solely by coding sequence abundance; it is modulated by untranslated regions, RNA secondary structure, chemical modifications, and 3′-end features that collectively shape ribosome recruitment and translational efficiency. DRS provides access to several of these regulatory layers simultaneously. DRS simultaneously resolves alternative PAS, 3′ UTR isoforms, and poly(A) tail length at the level of individual RNA molecules [52], [60]. APA remodels the 3′ UTR regulatory landscape, modulating access of RNA-binding proteins and microRNAs and thereby influencing mRNA stability, subcellular localization, and translational efficiency. In parallel, poly(A) tail length, often inversely correlated with steady-state mRNA abundance and sensitive to epitranscriptomic perturbations such as METTL3 depletion [60], directly shapes the efficiency with which transcripts are translated into protein. The 5′ and 3′ UTRs harbor powerful cis-regulatory elements that govern translational control. Studies using synthetic and native 5′ UTR libraries have demonstrated more than 100-fold variation in ribosome recruitment and translation initiation, driven by short sequence motif, RNA secondary structures, and upstream open reading frames (uORFs). Likewise, 3′ UTRs, particularly their structural features, exert strong influence over translation efficiency and serve as major hubs of post-transcriptional regulation. By preserving complete UTR context, DRS enables these regulatory features to be directly linked to isoform-specific proteomic output. m6A deposition within 3’ untranslated regions influences transcript abundance, 3’-end formation, and downstream phenotypes, such as circadian period regulation. Transcriptome-wide DRS analyses further reveal coordinated relationships among RNA methylation, stability, poly(A) tail dynamics, and AS, collectively reshaping translational efficiency and protein output. Transcriptome-wide RNA structure probing and modeling demonstrate that secondary structure, particularly within 5′ and 3′ UTRs, is a strong determinant of translational efficiency. Integrating such structural information with isoform-resolved DRS and quantitative proteomics enables direct evaluation of how defined RNA structural states modulate protein output.
Together, these advances position DRS as a central enabling technology for RNA-to-protein integration. By guiding proteogenomic database construction, improving isoform-resolved protein mapping, and capturing regulatory layers relevant for translation, DRS provides a unified framework for interpreting how complex transcriptomes give rise to functional proteomes. As proteomic depth and translatomic resolution continue to improve, DRS-based integration is expected to play an increasingly important role in elucidating post-transcriptional regulation and its contribution to disease mechanisms and therapeutic responses.
Multi-layer integration for regulatory network reconstruction
Beyond pairwise integration, the full potential of nanopore DRS emerges when it is integrated with multiple omics modalities to reconstruct RNA-centered regulatory networks (Figure 9E). Single-molecule DRS enables joint analysis of AS, epitranscriptomic decoration (such as m6A and m5C), RNA stability, and transcript abundance within the same read, revealing coordinated crosstalk among RNA regulatory layers that are otherwise analyzed in isolation [33], [39], [52], [60], [179]. Within such multi-layer frameworks, different omics modalities contribute complementary regulatory information, including chromatin accessibility defines transcriptional potential and regulatory input space, DRS captures mature transcript architecture and epitranscriptomic state, and proteomics quantifies protein output. Anchoring these layers on native RNA molecules enables mechanistic tracing of regulatory information flow from genome to phenotype. Empirical studies illustrate the power of this approach. In mouse organs subjected to fasting and feeding, integration of DRS with ATAC-seq and srRNA-seq uncovered thousands of previously unannotated transcript isoforms, along with tissue-specific shifts in poly(A) tail length and m6A patterns linked to metabolic pathways [367]. These findings demonstrate how chromatin accessibility and RNA processing are coordinated at the organ level to drive metabolic adaptation. Similarly, in bacterial and plant systems, integration of DRS-based modification calls with MeRIP-seq and Illumina RNA-seq has refined transcriptome-wide m6A maps and revealed relationships between RNA modification, operon organization, transcript abundance, and RNA decay dynamics [33], [52], [209]. Computational advances further extend these integrative frameworks. Deep learning-based models, such as SingleMod and pum6a, infer single-molecule RNA modification patterns directly from DRS signal data and use these features to explain effects on splicing, RNA stability, and stress-responsive gene expression programs, including those relevant to cancer [179], [405]. These approaches exemplify how DRS-derived chemical and structural information can be incorporated into predictive models that bridge RNA chemistry with gene expression phenotypes.
Across these applications, DRS plays a central role by providing molecular continuity between regulatory inputs and functional outputs. Because DRS preserves transcript identity across regulatory layers, it enables consistent mapping of information from DNA and chromatin state to RNA processing, translation, protein production, and metabolic outcome. This continuity is particularly valuable in complex regulatory scenarios where perturbations at one layer may be buffered, amplified, or rewired at another, obscuring causal relationships. Current multi-omics gene regulatory network (GRN) and enhancer-GRN frameworks typically integrate chromatin accessibility, transcription factor binding, transcriptomics, and occasionally proteomics to infer regulatory interactions. A recurring theme in these studies is the need to extend such models with additional molecular layers and more quantitative, causally informative measurements. DRS naturally fulfills this role by serving as the RNA continuity layer that connects chromatin and transcriptional regulation with downstream proteomic states. Incorporation of DRS into multi-omics network reconstruction therefore represents a key advance toward mechanistically grounded and predictive models of gene regulation.
Computational and analytical considerations
Effective multi-omics integration involving nanopore DRS critically depends on DRS-aware preprocessing and rigorous harmonization across a shared, isoform-resolved coordinate system (Figure 9F). Substantial differences in data resolution, noise characteristics, and sampling depth across omics modalities pose nontrivial analytical challenges. DRS data are inherently long-read and signal-rich, capturing molecule-level information on transcript structure, poly(A) tails, and RNA modifications, whereas most complementary omics assays, including srRNA-seq, ATAC-seq, ribosome profiling, and proteomics, produce fragmented or feature-based measurements. Bridging these fundamentally different data representations requires robust transcript annotation, accurate isoform quantification, and standardized feature mapping. Several characteristics of DRS complicate direct integration. Compared with short-read sequencing, DRS exhibits higher base-level error rates and generates a substantial fraction of truncated or non-full-length reads, which can obscure transcript boundaries and complicate isoform assignment and quantification [84], [405], [640]. These challenges are particularly pronounced in complex mammalian transcriptomes and for lowly expressed isoforms, where read coverage is sparse [672]. In addition, long-read datasets often show variability in TSS and PAS detection, making it difficult to define consistent transcript boundaries that are directly comparable across omics layers [646]. Limited sequencing throughput further constrains statistical power for differential analyses and complicates matching depth-rich short-read, ATAC-seq, or proteomics datasets [84], [86], [640]. Finally, DRS reports per-molecule signals and modification states, whereas most other omics data are summarized as aggregated genomic features, such as peak intensities or count matrices, creating an additional layer of abstraction that must be reconciled during integration [84], [614], [640], [665].
Robust, DRS-aware transcript annotation therefore represents a foundational design principle for harmonized multi-omics analysis. Long-read-optimized tools, including IsoQuant, Bambu, NAGATA, and SQANTI3, are essential for constructing high-quality isoform catalogs that accurately capture transcript structure, splice junctions, and transcript ends [84], [210], [646], [672]. Where possible, these annotations should be supported by orthogonal evidence from short-read RNA sequencing or specialized end-mapping assays. Hybrid short- and long-read pipelines are particularly effective, as they improve definition of transcript termini, strand assignment, and isoform structures, yielding a more reliable and biologically meaningful reference annotation [84], [613], [646]. This isoform catalog then serves as a common coordinate system onto which all other omics measurements can be projected. Accurate isoform quantification constitutes a second critical principle. DRS-specific quantification tools, such as NanoCount, IsoQuant, and NanoTrans modules, are designed to exploit long-read properties while filtering ambiguous alignments and partial reads [84], [86], [90], [206]. Benchmarking efforts, including the long-read RNA-seq genome annotation assessment project (LRGASP) framework, consistently demonstrate that reference-based approaches using high-quality isoform annotations yield the most reliable abundance estimates for integrative analyses [84]. Reliable quantification is essential not only for transcript-level comparisons but also for linking RNA features to downstream translation and protein output. A third principle is standardized coordinate and feature mapping across omics layers. Rather than collapsing measurements at the gene level, integration pipelines should map ATAC-seq peaks, chromatin immunoprecipitation sequencing (ChIP-seq) signals, ribosome footprints, proteomics peptides, and metabolite-associated genes onto the same isoform-resolved annotation [84], [614], [665]. This strategy fully leverages the strengths of DRS by preserving proteoform-specific and regulatory information that would otherwise be obscured by gene-level aggregation.
Collectively, integrative DRS-based multi-omics analyses require analytical pipelines that first address DRS-specific challenges in transcript annotation and quantification, and then systematically project all complementary omics data into a shared, isoform-resolved coordinate space. By doing so, these frameworks mitigate differences in resolution, noise structure, and sampling depth across data types, enabling coherent and mechanistically interpretable reconstruction of RNA-centered regulatory programs.