Introduction
With the accumulation of experimental and computational evidence, the multilayered and intricate roles of RNA in gene expression control have come into sharp focus. These roles span coding and non-coding RNAs, transcriptional and post-transcriptional regulation, as well as epigenetic modulation and RNA editing. Together, diverse RNA species form a finely tuned and highly dynamic regulatory network that safeguards the spatiotemporal precision and functional versatility of gene expression [1], [2]. In parallel, a growing repertoire of RNA modifications, typified by N6-methyladenosine (m6A), has emerged as key epitranscriptomic marks. Their contributions to transcript stability, splicing, nuclear export, translational efficiency, and RNA decay have been progressively elucidated [3], [4]. Analogous to DNA methylation and histone modifications, RNA modifications are highly dynamic and reversible, offering an additional regulatory layer that provides mechanistic insights into the precise control of gene expression programs [5], [6].
Over the past three decades, RNA sequencing technologies have evolved rapidly, driven by continual breakthroughs in sequencing platforms (Figure 1). This progression has moved from short-read RNA-seq (srRNA-seq) represented by Illumina platforms [7], to long-read cDNA sequencing represented by PacBio and Oxford Nanopore Technologies (ONT) [8], and, in the last decade, to the advent of nanopore direct RNA sequencing (DRS) [9]. Prior to the next-generation sequencing (NGS) era, Venter and colleagues in 1991 introduced and implemented the expressed sequence tag (EST) strategy, in which short cDNA fragments were sequenced to rapidly capture gene expression information [10]. Building on this, in 1997, Velculescu, Kinzler, and colleagues coined the term “transcriptome” and used ESTs to systematically catalog cellular transcripts, laying the conceptual foundation for modern transcriptomics [11]. The advent of RNA-seq marked a paradigm shift, freeing researchers from the limitations of single-gene, low-throughput approaches such as Northern blotting and microarrays, and enabling genome-wide quantification of transcript abundance [12]. Nevertheless, short-read sequencing is inherently limited by read length and the complexity of transcript reconstruction. In the context of long transcripts, intricate alternative splicing (AS) events, or tandem repetitive regions, assembly-based inference often fails to recover full-length structures at single-molecule resolution [13]. These limitations spurred the development of long-read cDNA sequencing, in which reverse-transcribed cDNAs, frequently enriched or amplified by reverse-transcription PCR (RT-PCR) or targeted capture, are sequenced in a single-molecule, enabling isoform-resolved transcriptome characterization [14].
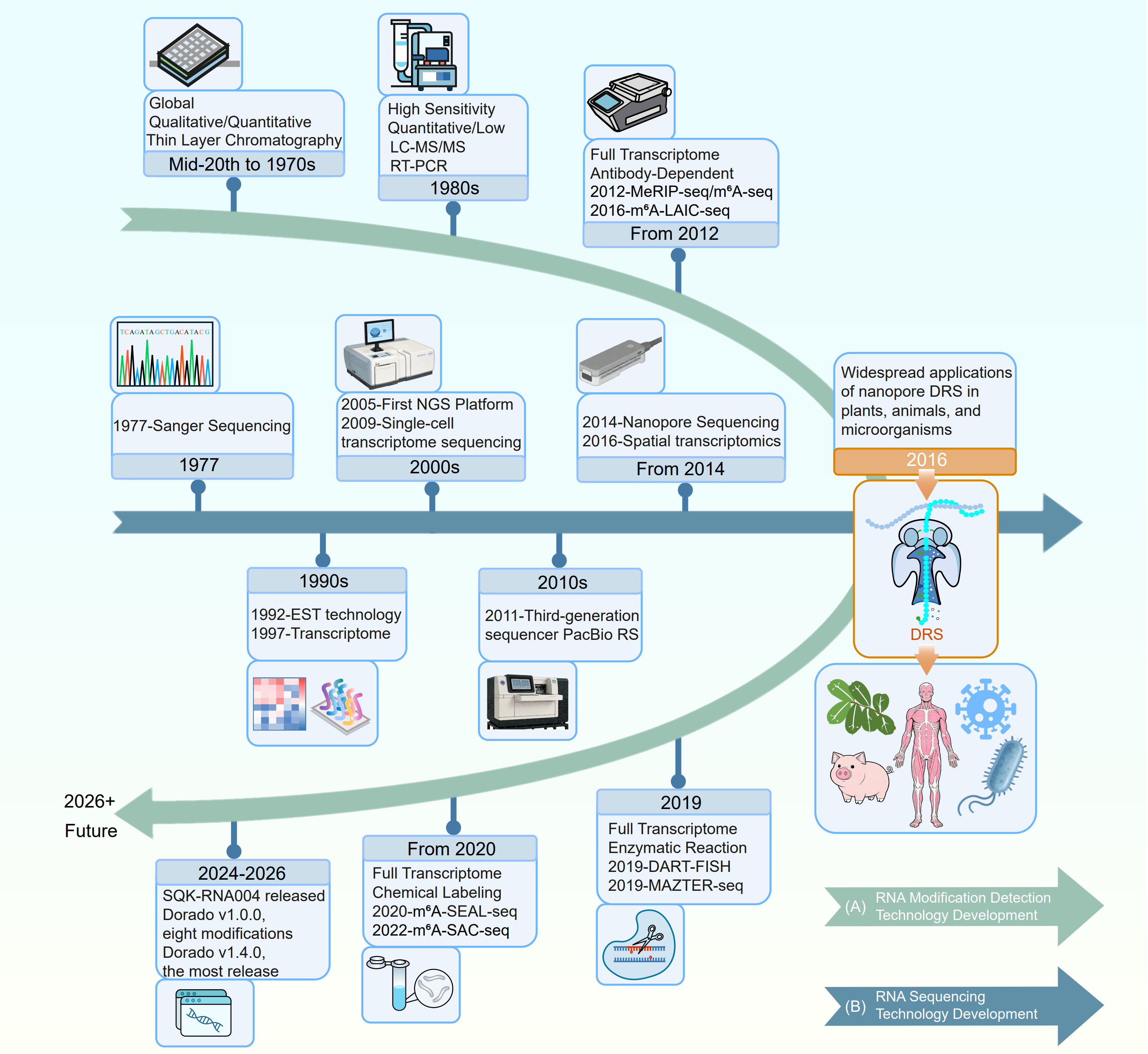
Figure 1. Timeline of major innovations in RNA sequencing and RNA modification technologies. Chronological overview of key methodological advances in (A) RNA modification detection and (B) RNA sequencing technologies. (A) Development of RNA modification analysis, from early qualitative and quantitative thin layer chromatography in the mid 20th century, through high sensitivity LC-MS/MS and RT-PCR-based approaches in the 1980s, to transcriptome wide antibody based mapping methods (e.g. MeRIP seq/m⁶A seq, m⁶A LAIC seq) established from 2012 onwards, together with more recent full transcriptome enzymatic and chemical labeling strategies (e.g. DART FISH, MAZTER seq, m⁶A SEAL seq, m⁶A SAC seq). Nanopore DRS with the SQK-RNA004 chemistry achieves enhanced performance and, when combined with Dorado software v1.0.0 and above, can detect up to eight distinct RNA modifications. (B) Evolution of RNA sequencing platforms, beginning with Sanger sequencing in 1977, followed by next generation sequencing (NGS) and single cell transcriptome sequencing in the 2000s, third generation long read platforms (e.g. PacBio RS) in the 2010s, and the advent of nanopore sequencing in 2014. The widespread application of nanopore direct RNA sequencing (DRS) to plant, animal and microbial transcriptomes from 2016 marks a key milestone, bridging RNA sequencing and epitranscriptomic profiling.
In parallel with sequencing advances, technologies for detecting and mapping RNA modifications have progressed from global quantification to nucleotide-level resolution. Early approaches, including paper chromatography, ion-exchange column chromatography, thin-layer chromatography (TLC), and dot-blot assays [15], [16], provided the first evidence for the existence and approximate abundance of major modifications such as m6A in eukaryotic mRNA. However, these methods lacked site specificity and sensitivity, limiting their utility for detailed mechanistic studies. Subsequent developments, such as high-performance liquid chromatography coupled with tandem mass spectrometry (LC-MS/MS), enabled femtomole-level quantification, while RT-PCR combined with Sanger sequencing allowed low-throughput validation of candidate sites. Nevertheless, LC-MS/MS does not retain transcript identity or positional information, and RT-PCR-based approaches are not scalable to transcriptome-wide analyses. With the advent of NGS, antibody-based methods such as m6A-seq and methylated RNA immunoprecipitation sequencing (MeRIP-seq) [17], [18] generated the first transcriptome-wide maps of RNA modifications by combining RNA fragmentation with immunoprecipitation. However, these approaches typically localize modifications only to enriched regions of about 100−200 nucleotides (nt), rather than to single bases. Crosslinking-enhanced strategies such as photo-crosslinking assisted m6A sequencing (PA-m6A-seq) and m6A individual-nucleotide-resolution cross-linking and immunoprecipitation (miCLIP) [19], [20], improved resolution to near single-base level but remain constrained by antibody specificity and potential false positives. Since 2018, non-antibody-based strategies have emerged, including chemical derivatization and reverse transcription signature-based approaches (e.g., Glyoxal and nitrite-mediated deamination of unmethylated adenosines sequencing (GLORI-seq) and m6A selective chemical labeling sequencing (m6A-SEAL-seq) [21], [22]), as well as enzyme-based methods such as enzyme-based m6A detection using MazF (MAZTER-seq) and deamination adjacent to RNA modification targets sequencing (DART-seq) [23], [24]. In parallel, related chemistries and workflows have been established for additional marks, including 5-methylcytosine (m5C) and pseudouridine (Ψ). Although these techniques have greatly improved resolution and quantitative comparability, they typically target specific modification types and require additional treatments. More fundamentally, most readouts are still anchored at the cDNA level, making it difficult to simultaneously resolve full-length transcript structures, modification landscapes, and poly(A) tail features at the single RNA molecule resolution.
A fundamental limitation shared by both short-read RNA-seq and long-read cDNA sequencing is that they sequence cDNA rather than native RNA molecules. This introduces three major sources of bias. First, they differ at the template level. In cDNA-based sequencing, reverse-transcribed products serve as the sequencing templates. Consequently, any biases introduced during RT, PCR amplification, or library preparation, such as template switching, 3′-end bias, GC-content-related bias, and amplification noise, are irreversibly embedded in the final sequencing data [25]. Second, the information content is inherently restricted. Conventional RNA-seq can only infer RNA modifications or poly(A) tail properties indirectly, for instance, by leveraging chemical or enzymatic enrichment or depth-of-coverage patterns to deduce a limited set of marks such as m6A or m5C [26], [27]. It cannot directly and simultaneously read “sequence, modification, and poly(A) tail” on the same individual molecule. Third, the observable molecular information remains incomplete. Transcripts truly exist as RNA molecules, whereas cDNA represents only a copied derivative. Under conditions such as disease, stress, or aging, RT efficiency can be differentially affected by complex secondary structures [28], [29] or modification-dense regions [30], [31], [32], leading to systematic under‑representation of biologically relevant transcripts [33]. DRS based on nanopore sequencing platforms overcomes these limitations by enabling single-molecule, long-read sequencing directly on native RNA templates. In this setup, RNA molecules are threaded through nanopores under the control of a motor protein. Their nucleotide sequence and modification status jointly shape the ionic current trace, which is then decoded by basecalling algorithms into sequence and can be further interrogated to infer modification events [9], [34]. More recently, the SQK-RNA004 chemistry, fully commercialized in 2024, delivers improved performance through redesigned nanopores and optimized motor proteins, and when paired with Dorado software starting from v1.0.0, it enables the identification of up to eight RNA modifications using nanopore DRS data. This technology thus provides new opportunities to investigate RNA molecules in their native biochemical and structural context.
Against this backdrop, one-shot measurement of “full-length transcript, poly(A) tail features, and RNA modifications” on the same molecule is rapidly emerging as a central goal for the next phase of RNA biology. Traditional multi-platform approaches struggle to meet the combined demands of throughput, cost, and quantitative consistency. DRS offers a conceptually appealing “one-shot, multi-layer readout” solution, creating new space for what can be regarded as a truly integrated “RNA-centric biology” that unifies the transcriptome, epitranscriptome, and poly(A) tail dynamics. Rather than viewing nanopore DRS as a replacement for existing transcriptomic methods, we position it as a complementary, molecule-resolved platform that provides an integrative entry point into the multiple layers of RNA regulation. In this Review, we therefore focus not only on what DRS can currently measure, but also on what still depends on model-based inference, which applications are best suited to DRS, and what forms of benchmarking and orthogonal validation are required to support different classes of biological conclusions. Building on the substantial body of methodological and applied studies on DRS published in recent years, we here provide a systematic overview of this technology, covering its physical principles, experimental features, cutting-edge applications, data-analysis strategies, and method development. Our goal is to connect the full pipeline “from biophysical principles to experimental design, from data processing to algorithmic innovation, and from basic discovery to translational applications”, thereby offering researchers in RNA biology a practically oriented and readily actionable review of DRS.